The positive effects from consuming several of the medicinal mushroom strains with people with Alzheimer, cognitive impairment, dementia and neurological diseases.
In Alzheimer’s disease, brain cells that process, store and retrieve information degenerate and die. Although scientists do not yet know the underlying cause of this destruction, they have identified several possible culprits.
One prime suspect is a microscopic brain protein fragment called beta-amyloid, a sticky compound that accumulates in the brain, disrupting communication between brain cells and eventually killing them. Some researchers believe that flaws in the processes governing production, accumulation or disposal of beta-amyloid are the primary cause of Alzheimer’s. This theory is called “the amyloid hypothesis.”
There is an elevated increase in ATP in the body when consuming the mushroom strains. ATP is an important communication and energy molecule in each cell. When we age the level or ATP levels in our cells are reduced. Consuming the mushroom strains increases ATP. This improves the communication between our cells resulting in the proliferation of the needed T cells to remove damaged, mutated and cancerous cells and for the proliferation of your own stem cells to repair and build new cells.
In anecdotal tests with patients that had lost memory when consuming the mushroom product over a few weeks with a dosage of 16 grams (8 teaspoons) or more a day, they saw their memories started to return and remain. A large study is planned to determine mechanisms and pathways and dosage observations to design the appropriate protocols.
In the International Journal of Medicinal Mushrooms, Dr. Takashi Mizuno of Shizouka University in Japan studies we done when used for the treatment of Alzheimer’s disease. The disease is caused in the brain by plaque buildup around nerve cells and by tangled up nerve fibers call neurofibrillary tangles. Compounds in the mushroom strain is useful in the treatment of Alzheimer’s disease.
Low levels of nerve growth factor (NGF) are seen in Alzheimer’s disease with implications for the application of the Hericium strain in the management of this condition. In addition, synaptic degeneration, with loss of synaptic density proteins, has been shown to be a key mode of neurodegeneration in Alzheimer’s disease with beta-amyloid (Abeta) identified as a cause of synaptic dysfunction and contributor to Alzheimer’s disease pathology. The Ganoderma strain of mushrooms we use traditionally is used for antiaging properties, has been shown to significantly inhibit Abeta-induced synaptotoxicity and also inhibit Abeta-triggered DEVD cleavage in a dose-dependent manner with potential clinical application in management of Alzheimer’s disease and a mushroom strain considered to have anti-ageing properties, has been shown to inhibit the enzyme associated with release of Abeta. Amyloid beta denotes peptides of 36–43 amino acids that are crucially involved in Alzheimer's disease as the main component of the amyloid plaques found in the brains of Alzheimer patients. The peptides derive from the amyloid precursor protein, which is cleaved by beta secretase and gamma secretase to yield Aβ.
Aβ molecules can aggregate to form flexible soluble oligomers which may exist in several forms. It is now believed that certain misfolded oligomers can induce other Aβ molecules to also take the misfolded oligomeric form, leading to a chain reaction akin to a prion infection. The oligomers are toxic to nerve cells. The other protein implicated in Alzheimer's disease, tau protein, also forms such prion-like misfolded oligomers, and there is some evidence that misfolded Aβ can induce tau to misfold.
Dr. Joyce Czop of Harvard University wrote that Beta Glucans from various mushroom strains are pharmacologic agents that rapidly enhance the host resistance to a variety of biologic insults through mechanism involving macrophage activation.
Recent studies on Schwann cells (neurolemmocytes are the principal glia of the peripheral nervous system (PNS)), at the neuromuscular junction and non-synaptic regions of premyelinated axons indicate that extracellular ATP can act as an activity-dependent signaling molecule in communication between neurons and glia. Several mechanisms have been observed for the regulated release of ATP from synaptic and non-synaptic regions, and a diverse family of receptors for extracellular ATP has been characterized. The findings suggest functional consequences of neuron–glial communication beyond homeostasis of the extracellular environment surrounding neurons, including regulating synaptic strength, gene expression, mitotic rate, and differentiation of glia according to impulse activity in neural circuits.
Scientists have discovered an interesting compound in the strains of the medicinal mushroom Hericium, Gonoderma, Agarius and Cordyceps, erinacine, found to be a kappa opiod receptor-biding inhibitor. These anti-convulsants that can be neuro-protective in epilepsy and stroke, as well as brain and spinal cord injuries.
There is a chemical called Cyathan which is a nerve growth stimulator in these mushroom strains we use.
Son et al. (2006) found water extracts of this strain of this specie of mushroom induced iNOS gene expression followed by NO (Nitric oxide) production in macrophages via enhancing the activation of transcription factor NFkappaB.
A study of one hundred patients in a rehabilitation hospital in Japan looked at the effect of five grams (2 teaspoons) of this mushroom strain or a placebo in their soup for six months. These patients were elderly and suffered from cerebrovascular disease, Degenerative othroppedic disease, Parkinson’s disease, Alzheimer’s, Spino-cerebellar degeneration, Diabetic neuropathy, Spinal cord injury, or Disuse syndrome. Those consuming the product saw a cognitive and comprehension improvement.
A study of twenty-nine men and women aged fifty to eighty with mild cognitive problems was conducted by Mori et al. (2008). This double-blind, placebo-controlled trial showed significant improvement.
One study found in vitro evidence of myelin-generating effect on nerve and cerebellar glia cells.
Works by Moldavan et al. (2007) found extracts exerted neurotropic action and improved the myelination process in maturing fibers, did not affect nerve cell growth in vitro, and did not exert a toxic effect or nerve cell damage.
The compound 3-hydroxy-hericenone F extracted from this mushroom identified by Ueda et al. (2008) shows protection against endoplasmic reticulum stress-dependent Neuro2 cell death.
Nagai et al. (2006) identified di-linoleotl-phopshatydl-ethanolamine as a compound the reduced ER stress and protects neuronal cells.
The Cordycep strain used in our product is considered to be a medicinal mushroom in classical Asian pharmacology, as well as traditional Chinese and Tibetan medicines. The Pharmaceutical Society of Japan’s Biological and Pharmaceutical Bulletin recently reported the mushroom to be valuable for protection against Alzheimer’s as it prevents neuronal cell death and memory loss through its antioxidant and anti-inflammatory effects.
Other studies proved that Cordyceps is non-toxic and promotes cognitive health.
Neuroprotection refers to the mechanisms and strategies used to protect against nerve injury or degeneration and to prevent the breakdown of the central nervous system.
Researchers are looking for ways protect the body after acute events, such as stroke or nervous system injury, and to help with chronic nervous system diseases such as Alzheimer's, Parkinson's or multiple sclerosis (MS).
The development of neuroprotective agents is still underway, although there are some in use today.
Current neuroprotectors cannot reverse the damage already done, but they may protect against further nerve damage and slow down any degeneration, or breakdown, of the central nervous system (CNS).
Scientists are currently investigating a wide range of treatments. Some products can potentially be used in more than one disorder, as different disorders share many of the underlying mechanisms.
Ma, Bing-Ji, Jin-Wen Shen, Hai-You Yu, Yuan Ruan, Ting-Ting Wu & XU Zhao, 2010. “Hericenones and erinacines: stimulators of nerve growth factor (NGF) Biosyntheses in Hericium erinaceus.” Mycology: An International Journal on Fungal Biology. 1(2): 92-98.
Mori, K., Inatomi, S., Ouchi, K. Azumi, Y and Tuchida T. 2009. “ Improving effects of the mushroom Yamabushitake (Hericium erinaceus) on mild cognitive impairment: a double blinded, placebo controlled clinical trial “Phytother Res. 23:367-372.
Thal, L.J. Kantarci, K., Reiman, E.M., Klunk, W.E., Weiner, M.W., Zetterberg, H., Galasko, D., Pratico, D., Griffin, S., Schenk, D., Siemers, E. 2006. “The role of biomarkers in clinical trials for Alsheimer disease” 20 (1):6-15.
Mori, K., Obara, Y., Moriya, T., Inatomi, S., Nakahata, N. 2011. “Effects of Hericium erinaceus on amyloid β (25-35) peptide-induced learning and memory deficits in mice.” Biomed Res. 31 (4):231-7.
_______________________________________________________________________________________________________
Kappa Opioids, Salvinorin A and Major Depressive Disorder.
Taylor GT, Manzella F1.
Author information
Abstract
Opioids are traditionally associated with pain, analgesia and drug abuse. It is now clear, however, that the opioids are central players in mood. The implications for mood disorders, particularly clinical depression, suggest a paradigm shift from the monoamine neurotransmitters to the opioids either alone or in interaction with monoamine neurons. We have a special interest in dynorphin, the last of the major endogenous opioids to be isolated and identified. Dynorphin is derived from the Greek word for power, dynamis, which hints at the expectation that the neuropeptide held for its discoverers. Yet, dynorphin and its opioid receptor subtype, kappa, has always taken a backseat to the endogenous b-endorphin and the exogenous morphine that both bind the mu opioid receptor subtype. That may be changing as the dynorphin/ kappa system has been shown to have different, often opposite, neurophysiological and behavioral influences. This includes major depressive disorder (MDD). Here, we have undertaken a review of dynorphin/ kappa neurobiology as related to behaviors, especially MDD. Highlights include the unique features of dynorphin and kappa receptors and the special relation of a plantbased agonist of the kappa receptor salvinorin A. In addition to acting as a kappa opioid agonist, we conclude that salvinorin A has a complex pharmacologic profile, with potential additional mechanisms of action. Its unique neurophysiological effects make Salvinorina A an ideal candidate for MDD treatment research.
PMID:
26903446
PMCID:
PMC4825947
________________________________________________________________________________________________
NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review
Ruey-Horng Shih,1 Chen-Yu Wang,2 and Chuen-Mao Yang2,*
Author information ► Article notes ► Copyright and License information ► Disclaimer
This article has been cited by other articles in PMC.
Abstract
The NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells) transcription factor family is a pleiotropic regulator of many cellular signaling pathways, providing a mechanism for the cells in response to a wide variety of stimuli linking to inflammation. The stimulated cells will be regulated by not only the canonical but also non-canonical NF-κB pathways. To initiate both of these pathways, IκB-degradation triggers NF-κB release and the nuclear translocated-heterodimer (or homodimer) can associate with the κB sites of promoter to regulate the gene transcriptions. NF-κB ubiquitously expresses in neurons and the constitutive NF-κB activation is associated with processing of neuronal information. NF-κB can regulate the transcription of genes such as chemokines, cytokines, proinflammatory enzymes, adhesion molecules, proinflammatory transcription factors, and other factors to modulate the neuronal survival. In neuronal insult, NF-κB constitutively active in neuron cell bodies can protect neurons against different injuries and regulate the neuronal inflammatory reactions. Besides neurons, NF-κB transcription factors are abundant in glial cells and cerebral blood vessels and the diverse functions of NF-κB also regulate the inflammatory reaction around the neuronal environment. NF-κB transcription factors are abundant in the brain and exhibit diverse functions. Several central nerve system (CNS) diseases are linked to NF-κB activated by inflammatory mediators. The RelA and c-Rel expression produce opposite effects on neuronal survival. Importantly, c-Rel expression in CNS plays a critical role in anti-apoptosis and reduces the age-related behaviors. Moreover, the different subunits of NF-κB dimer formation can modulate the neuroninflammation, neuronal protection, or neurotoxicity. The diverse functions of NF-κB depend on the subunits of the NF-κB dimer-formation which enable us to develop a therapeutic approach to neuroinflammation based on a new concept of inflammation as a strategic tool in neuronal cells. However, the detail role of NF-κB in neuroinflammation, remains to be clarified. In the present article, we provide an updated review of the current state of our knowledge about relationship between NF-κB and neuroinflammation.
Keywords: NF-kappaB, neuroinflammation, neuroprotection, adhesion molecules, proinflammatory transcription factors
NF-κB Family Members and Disease Control
NF-κB exerts effects on almost all cell types in the body, playing an important function in inflammation, immune responses, cell cycle, and cell survival (Sen and Baltimore, 1986; Li and Verma, 2002; Kaltschmidt et al., 2005; Mattson, 2005; Ledoux and Perkins, 2014). NF-κB has been recognized as a member of Rel family of transcription factors. In mammals, there are five different members to compose the NF-κB family: p65 (RelA), RelB, c-Rel, p50/p105 (NF-κB1), and p52/p100 (NF-κB2) which have the similar amino acid sequence, the RHD (Rel homology domain, over approximate 300 amino acids) of these proteins (Chen and Greene, 2004). The activated NF-κB subunits will assemble to form the homo-or hetero-dimerized transcription factor complexes displaying the DNA-binding ability and transactivation potentials. The most widely studied form of NF-κB is a heterodimer of the p50 and p65 subunits and is a potent activator of gene transcription (Schmitz and Baeuerle, 1991). NF-κB is activated by a wide variety of agents including viruses, bacterial toxins such as lipopolysaccharide (LPS), UV light, oxidative stresses such as free radicals and cigarette smoke, inflammatory stimuli, cytokines, carcinogens, tumor promoters, and various mitogens (Baeuerle and Henkel, 1994; Baldwin, 1996). NF-κB regulates the expression of almost 500 different genes, including enzymes [e.g., cyclooxygenase (COX)-2, 5-lipoxygenase (LOX), and inducible NO synthase (iNOS)], cytokines [such as interleukin (IL)-1, IL-6, IL-8, chemokines, and tumor necrosis factor (TNF)], adhesion molecules, cell cycle regulatory molecules, and angiogenic factors (Duh et al., 1989; Kaltschmidt et al., 1993; Ahn and Aggarwal, 2005; Gupta et al., 2010a,b). The activation of NF-κB, especially the constitutively activated NF-κB in chronic inflammatory patients, has been found the critical linkage with a wide variety of human diseases, including asthma, atherosclerosis, AIDS, Alzheimer’s disease (AD), Parkinson’s disease (PD), rheumatoid arthritis, cancer, diabetes, and osteoporosis which belong to autoimmune/inflammatory diseases (Vallabhapurapu and Karin, 2009; Gupta et al., 2010b). The opposite, several native or artificial agents such as Th2 cytokines (IL-4, IL-13, and IL-10), interferons, endocrine hormones (LH, HCG, MSH, and GH), phytochemicals, corticosteroids, and immunosuppressive drugs, are known to block the specific signaling transductions and suppress NF-κB activation (Ahn and Aggarwal, 2005). Therefore, regulation and dysregulation of NF-κB play a key role in diseases control.
The Canonical and Non-Canonical NF-κB Signaling Pathways
Based on the previous studies, NF-κB is activated via two distinct kinase-dependent pathways, the classical/canonical NF-κB pathway and the alternative/non-canonical NF-κB pathway. The most extensively studied NF-κB activation pathway is the canonical pathway (Figure1, modified from Noort et al., 2015), which can be mediated through activation of a variety of cell surface receptors, including IL-1 receptor, Toll-like receptors (TLRs), and TNF receptor, in response to pro-inflammatory mediators like IL-1, LPS, and TNF, as well as via triggering of the T-cell receptor or B-cell receptor. The inactive NF-κB resides in the cytoplasm and associates or links with the natural biological inhibitor IκB. The NF-κB function and nuclear translocation ability are sequestered in the cytoplasm and nuclear compartments, respectively (Verma et al., 1995; Baeuerle and Baltimore, 1996). The IκB family members include IκBα, IκBβ, p105/IκBγ (precursor of p50), p100 (precursor of p52), and IκBε (Li and Nabel, 1997; Whiteside et al., 1997). Each shares a series of ankyrin repeats which sequester NF-κB in the cytosol by masking its nuclear localization signal (NLS) and also prevents NF-κB from binding to DNA by masking its DNA binding domain. Treatment of cells with various stimuli activates IκB kinase complex, for example, leading to the phosphorylation of serines 32 and 36 of IκBα or serines 19 and 23 of IκBβ (DiDonato et al., 1997; Mercurio et al., 1997; Regnier et al., 1997; Zandi et al., 1997). These phosphorylation events target IκB for ubiquitin-dependent degradation through the 26S proteasome complex, resulting in the release and nuclear translocation of NF-κB (Finco and Baldwin, 1995; Thanos and Maniatis, 1995). Briefly, NF-κB is expressed ubiquitously in the cytoplasm of almost all cell types. The activated NF-κB will translocate from cytoplasm to nucleus and the NF-κB-dimer can bind to the κB site of promoter. In this classical pathway, inhibitor of κB kinase (IKK)β is required for NF-κB activation (Tak et al., 2001), whereas IKKα is redundant (Vallabhapurapu and Karin, 2009). However, the canonical NF-κB pathway is essential for both acute and chronic inflammatory responses. Moreover, this pathway is implicated in cell proliferation and survival, demonstrated by constitutively active NF-κB signaling in many tissues (Ben-Neriah and Karin, 2011).
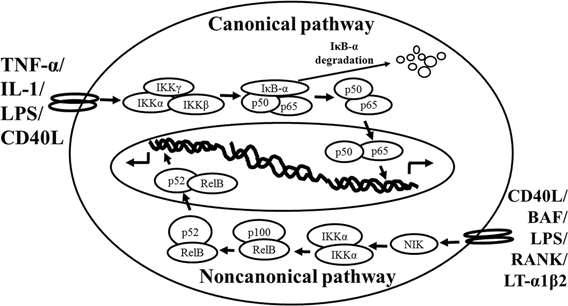
Schematic representation of the canonical and non-canonical nuclear factor (NF)-κB activation pathways. The canonical NF-κB pathway (upper) can be activated by a wide range of various stimuli, including tumor necrosis factor (TNF)-α, interlukin (IL)-1, lipopolysaccharide (LPS), and Toll-like receptors ligand (such as CD40L). Initiation of the canonical pathway via Toll-like receptor or cytokine receptor signaling depends on the inhibitor of κB kinase (IKK) complex, which is composed of the kinases IKKα and IKKβ, and the regulatory subunit IKKγ (NEMO). Activated IKK phosphorylates the inhibitory subunit IκBα leading to its degradation. The released NF-κB dimers (p50-p65) translocate to the nucleus and bind to κB site of chromosome to induce transcription of NF-κB targeted genes. The non-canonical pathway (lower) is activated by specific stimuli including B cell activating factor (BAF) belonging to the TNF family receptor, LPS, lymphotoxin (LT) α1β2, receptor activator of NF-κB (RANK), and CD40L. NF-κB inducing kinase (NIK) is stabilized. When stimulated, NIK is activated and recruits IKKα to the p100 complex to phosphorylate p100, leading to p100 ubiquitination. P52, the processing product of p100, generates the activated p52/RelB NF-κB complex, which is able to translocate to the nucleus and induce the downstream gene expressions.
The non-canonical NF-κB pathway (Figure1), can be triggered by the activation of members of the TNF-receptor superfamily including B cell activating factor (BAF), belonging to the TNF family receptor, CD40, lymphotoxin β (LTβ) receptor, and receptor activator of NF-κB (RANK). Of note, these receptors trigger not only the non-canonical NF-κB pathway, but also the canonical pathway, simultaneously. The non-canonical NF-κB pathway is strictly dependent on IKKα homodimers and unlike the canonical pathway, the IKKβ or IKKγ is not involved in the IκB phosphorylation (Sun, 2012). To regulate the non-canonical pathway, expression of NF-κB inducing kinase (NIK) plays a role as the most important kinase. In the steady state, TNF receptor-associated factor (TRAF)3 mediates recruitment of NIK to TRAF2, which leads to NIK ubiquitination and continuous degradation. Consequently, endogenous levels of NIK are very low and the NF-κB complex is retained in the cytoplasm and kept inactive. Upon activation of the non-canonical NF-κB pathway, TRAF2 induces proteolysis of TRAF3. Degradation of TRAF3 prevents targeting of newly synthesized NIK, resulting in NIK release and accumulation. Subsequently, NIK induces p100 phosphorylation by IKKα homodimers and partial degradation to release p52. Next, mainly p52-RelB heterodimers translocate to the nucleus, leading to transcription of target genes. Whereas canonical NF-κB activation is rapid and independent of protein synthesis, non-canonical NF-κB activation requires NIK synthesis and accumulation. Consequently, the kinetics of this pathway are considerably slower (Vallabhapurapu and Karin, 2009; Sun, 2012).
There are cross-talks between these two pathways. IKKα has, for instance, been described to also have nuclear functions and serve as a regulator of canonical NF-κB-dependent gene expression through control of promoter-associated histone phosphorylation exposed to cytokines (Anest et al., 2003; Yamamoto et al., 2003). It has been demonstrated that the activated canonical pathway not only initiated the signal transduction of NF-κB but also suppressed basal non-canonical signaling in immune cells (Gray et al., 2014). Interestingly, under certain circumstances and other stimuli (including TNF) can also activate non-canonical NF-κB signaling in specific cell types (Zhang et al., 2014), and IKKα is critical for interferon-α production induced by TLR 7 and 9 (Hoshino et al., 2006).
The NF-κB Family Members in the Brain Location
The expression of NF-κB transcription factors is abundant in the brain. The basal levels of NF-κB expression have been identified in the brain where their amounts are higher than those of peripheral tissues. Several lines of evidence indicate that constitutively activated NF-κB is found in glutamatergic neurons of the central nervous system (CNS), such as cerebral cortex (layers 2, 4, and 5) and hippocampus (granule cells and pyramidal neurons of CA1 and CA3; Kaltschmidt et al., 1993, 1994, 1995). A number of studies also show constitutive NF-κB activity in various rodent brain regions such as amygdala, cerebral cortex, cerebellum, hippocampus, hypothalamus, and olfactory lobes (Schmidt-Ullrich et al., 1996). Among members of NF-κB, all of the complexes of c-Rel/p65, p50/p65 heterodimer, and p50 homodimers are detected in the developing rat brain (Bakalkin et al., 1993). While to analyze the distribution of NF-κB, the released p65 and p50 NF-κB subunits are abundantly expressed in neurons. Moreover, p50/p65 heterodimers are located in the cell nucleus and exhibit constitutive activity in the adult brain (Kaltschmidt et al., 2005; Meffert and Baltimore, 2005). In the developed rodent brain, the p50/p65 heterodimeric variant of NF-κB is converted to the major κB-binding complex (Schmidt-Ullrich et al., 1996; Meffert et al., 2003). It is important for the neuronal physiological characteristics, for example, constitutive NF-κB activity in glutamatergic neurons of the hippocampus and cerebral cortex can be suppressed by N-methyl-D-aspartate, and to a lesser extent AMPA, glutamate receptor antagonists, as well as L-type Ca2+ channel blockers (Lilienbaum and Israel, 2003; Meffert et al., 2003). These studies suggest that constitutive NF-κB activity is modulated by physiological basal synaptic transmission. However, inducible NF-κB is detected in synapses, glutamatergic stimulation activates retrograde transport of p65 protein from synapses to the cell nucleus (Kaltschmidt et al., 1993; Meberg et al., 1996; Meffert et al., 2003). Thus, NF-κB is involved in translation of short-lasting synaptic signals to persistent changes in gene expression (Wellmann et al., 2001; Meffert et al., 2003). The activated IKK ant it’s product, phosphorylated IκBα, were detected within the axon initial segment, the site where action potentials are generated (Schultz et al., 2006), suggesting that constitutive NF-κB activation is involved in the processing of neuronal information.
NF-κB and Neuroinflammatory Mediators
At the molecular level, inflammation is regulated by numerous molecules and factors, including adhesion molecules [intercellular adhesion molecule (ICAM-1), vascular cell adhesion molecule (VCAM)-1, endothelial-leukocyte adhesion molecule (ELAM)-1], chemokines (such as monocyte chemoattractant protein 1, IL-8), cytokines (IL-1, IL-2, IL-6, IL-12, TNF-α, TNF-β), signal transducer and activator of transcription (STAT)-3, proinflammatory enzymes [COX-2, 5-LOX, 12-LOX, matrix metalloproteinases (MMPs), prostate-specific antigen (PSA), C-reactive protein], vascular endothelial growth factor (VEGF), and proinflammatory transcription factors NF-κB (Aggarwal, 2004). Among these mediators, NF-κB is the central regulator of inflammation (Lukiw and Bazan, 1998; Aggarwal, 2004; Ahn and Aggarwal, 2005). For example: IL-1β treatment can induce COX-2 expression in canine tracheal smooth muscle cells (Yang et al., 2002) and ICAM-1 expression in human rheumatoid arthritis synovial fibroblasts (Yang et al., 2010), respectively. LPS, to mimic the bacterial infection, and endothelin-1 also can induce COX-2 and PGE2 expression in mouse brain microvascular endothelial (bEnd.3) cells (Shih and Yang, 2010; Lin et al., 2013). TNF-α can induce ICAM-1 expression in osteoblast-like MC3T3-E1 cells (Tsai et al., 2014). All of these target proteins syntheses are mediated through NF-κB-dependent signaling pathway.
NF-κB has been shown to activate more than 500 genes, which are implicated in inflammation related responses (Gupta et al., 2010a,b). The NF-κB family is suggested to be the most extensively studied target in inflammation issue for its critical role (Chen and Greene, 2004; Lin and Karin, 2007). In neuroinflammation, NF-κB can be transiently activated by various stimuli, like acute alcohol exposure, which induces neuroinflammatory responses in mice (Yakovleva et al., 2011). The role of NF-κB is critical in the regulation of neuroinflammation-associated disease pathogenesis (Niranjan, 2013).
NF-κB: A Neuroprotective Role or a Neurotoxic Role
In the CNS, NF-κB transcription factors are key players in a number of physiological processes such as neurogenesis (Koo et al., 2010), neuritogenesis (Rolls et al., 2007), and synaptic plasticity which related to learning and memory (Levenson et al., 2004; O’Riordan et al., 2006; Ahn et al., 2008). A number of studies also provide evidence that activation of NF-κB protects neurons against the different injuries such as excitotoxicity (Mattson, 2005), and oxidative stress (Sarnico et al., 2009b), as well as amyloid β peptide toxicity (Barger et al., 1995; Kaltschmidt et al., 1997) and exerts as a cellular defense program. Apoptotic cortical neurons have been observed to be rescued by overexpression of p65, while enhanced damage by IκB super-repressor or dominant negative NF-κB-inducing kinase (NIK; Bhakar et al., 2002). NF-κB is constitutively active, and involved in neuronal injury as well as neuroprotection in neuron cell bodies, however, NF-κB is present in a latent form at the synapse. Only when NF-κB is activated, it can be transported to the neuron cell nucleus (Yakovleva et al., 2011).
Besides neurons, the roles of NF-κB in astroglia/microglia have been studied in relation to brain injury (O’Neill and Kaltschmidt, 1997; Block et al., 2007; Kaltschmidt and Kaltschmidt, 2009). Briefly, NF-κB is present in a latent form in glia of naive animals (Schmidt-Ullrich et al., 1996; Bhakar et al., 2002). NF-κB may be activated under pathological conditions such as exposure to HIV-1 Tat or amyloid β peptide (Aβ) leading to the production of nitric oxide (Akama et al., 1998; El-Hage et al., 2008). It has been shown that glia responses to injury triggered by endogenous ligands for TLR and TLR signaling are mediated through the NF-κB (Akira and Takeda, 2004). Moreover, inhibition of astroglial NF-κB signaling leads to reduced chemokine expression and leukocyte entry into the injured CNS (Brambilla et al., 2005; Khorooshi et al., 2008). NF-κB has been shown to play the regulatory role of astrocytes on immune and inflammatory responses (Farina et al., 2007). Microgliosis is a common pathologically neurodegenerative disorder. Microglial activation of NF-κB plays a central role associated with the release reactive oxygen species and proinflammatory cytokines (such as IL-1β, interferon-γ, and TNF-α) that can cause secondary neurotoxicity (Kaltschmidt et al., 1993; Block et al., 2007). Briefly, in glia, NF-κB is inducible and regulates inflammatory processes that exacerbate inflammation-induced neurodegeneration (Yakovleva et al., 2011). NF-κB has been also demonstrated as a major signal transducer affecting cellular permeability, endocytosis, and intracellular trafficking at the level of the blood–brain barrier (Stone et al., 2011). Activation of NF-κB signaling by LPS has been shown to induce inflammatory target protein COX-2 and PGE2 production leading to cerebral vascular inflammation (Pan et al., 2010; Shih and Yang, 2010). All of above studies show that NF-κB transcription factors are abundant in the brain where they have diverse functions among neurons, glia, and cerebral blood vessels.
Effects of NF-κB on Inflammatory-Associated with Pain
Constitutive activation of NF-κB is detected mostly in glutamatergic neurons. NF-κB in glia has a lower basal activity and is highly inducible, which plays a crucial role in brain inflammation (Kaltschmidt and Kaltschmidt, 2009). A role of glial NF-κB in pain research has attracted more attention. Pain signaling can arise from the activation of specific high-threshold PNS neurons (nociceptors) and could serve as a sensing mechanism to prevent further injury. In clinic, pain signaling can arise not only from damage to the nervous system (neuropathic pain), but also from chronic inflammation (inflammatory pain). Interestingly, an impairment of acute and inflammatory nociception has been revealed in p50-/- mice in a previous study (Niederberger et al., 2007). Moreover, inhibition of astroglial NF-κB can reduce inflammation and therefore improve functional recovery after spinal cord injury (Brambilla et al., 2005). All of these data suggests that NF-κB plays a crucial role on inflammatory pain in CNS.
Different NF-κB Complexes Differentially Regulate Neuronal Survival in Brain Damage: p50/RelA vs. p50/c-Rel
In recent years, NF-κB dysregulation has been shown to link to neurodegenerative mechanisms that occur in brain during trauma or ischemia (Bethea et al., 1998; Schneider et al., 1999), as well as in the brain of patients suffered by PD (Hunot et al., 1997; Ghosh et al., 2007) or AD (Boissiere et al., 1997; Kaltschmidt et al., 1997; Lukiw and Bazan, 1998). These CNS diseases are associated with neuroinflammatory mediators. More evidence has shown that the neuronal response to external stimuli relies on a differential activation of NF-κB dimers. RelA or c-Rel expression produces opposite effects on neuron survival (Pizzi et al., 2002, 2005b; Sarnico et al., 2009b).
Among the members of NF-κB, the RelA subunit, composing the activated p50/RelA dimer, and its post-transcriptional modifications play a pivotal role in the onset of neurodegenerative processes triggered by ischemic insults (Inta et al., 2006; Sarnico et al., 2009a,b) as well as glutamate (Pizzi et al., 2002) or Aβ toxicity (Pizzi et al., 2005b; Inta et al., 2006; Lanzillotta et al., 2010). In ischemic stroke, activated RelA induces the expression of the 1B isoform of the divalent metal transporter-1(1B/DMT1) which can exert as an upstream response for iron accumulation and contributing to neuronal cell death after injury (Ingrassia et al., 2012). Notably, RelA is demonstrated as a most contributing subunit in degenerative changes associated with senescence in a mice model (Tilstra et al., 2012).
RelA has been demonstrated to contributing to neuronal cell death, while the overexpression of c-Rel factor can limit the cell death. The c-Rel factor is reduced in neurons exposed to oxygen–glucose deprivation (OGD), interestingly, the overexpression of c-Rel prevents neuronal loss in cortical neurons exposed to OGD. This protective effect involves in increasing the transcription of Bcl-xL gene (Pizzi et al., 2009; Sarnico et al., 2009a,b). Similarly, knocking down c-Rel expression exacerbated neuronal susceptibility to OGD-mediated damage (Pizzi et al., 2009). Further, knocking out c-Rel expression appeared insensitive to neuroprotective activity of leptin, a c-Rel inducer capable to limit cortical damage in wild-type mice and mice brain ischemia (Valerio et al., 2006, 2009). Therefore, the c-Rel subunit within activated NF-κB dimers also counteracts the ischemic injury acting as an innate mechanism of neuroprotection (Sarnico et al., 2009a,b). In addition, overexpression of c-Rel in cultured neurons promotes anti-apoptotic effects by inducing the transcription of manganese superoxide dismutase (MnSOD; Chen et al., 2000; Bernard et al., 2001; Pizzi et al., 2005a). On the viewpoint of disease events, the deficiency of c-Rel induces an age-related behavioral Parkinsonism in mice, with degeneration of nigral dopaminergic (DA) neurons and development of a PD-like neuropathology (Baiguera et al., 2012). Recent evidence has shown that activation of NF-κB drives the systemic and brain aging processes in mice (Adler et al., 2007; Zhang et al., 2013). In brain ischemic tissue of mice subjected to permanent middle cerebral artery occlusion (MCAO) and in primary cortical neurons exposed to OGD, NF-κB followed a pattern of increasing p50/RelA dimmer (Crack et al., 2006; Inta et al., 2006) and decreasing c-Rel-containing dimmers (Sarnico et 12
al., 2009b). Inhibition of c-Rel-containing dimers and activation of p50/RelA are key events in the pathogenesis of brain injury. These data strongly suggested that NF-κB transcription factors have diverse functions that depend on the composition of the NF-κB complex (Lanzillotta et al., 2015).
Conclusion
The role of NF-κB is critical in the regulation of neuroinflammation-associated disease pathogenesis. NF-κB transcription factors are abundant and constitutive activation in brain where they have diverse functions among neurons, glia, and cerebral blood vessels. These functional diversions are dependent on the recruitment of components of the NF-κB dimer formation. Especially, c-Rel containing NF-κB dimers can induce the Bcl-xL and MnSOD expression and exert as anti-apoptotic effects while the pro-apoptotic effect elicited by NF-κB p50/RelA dimer. The imbalance of NF-κB dimer formation between RelA and c-Rel might result in the pathological process in certain neurons. The roles of NF-κB in neurological damage have been illustrated in Figure 2. The detail signal transduction pathways in different compositions of the NF-κB complex remain to be clarified. Obviously much more work is required to elucidate the role of NF-κB in the neuroinflammatory signaling pathways, which in turn will enable us to devise a therapeutic approach to neuroinflammation based on a new concept of inflammation as a strategic tool by which inflammatory neuronal cells can be made more susceptible to drugs than normal cells. By understanding the signal transduction pathways mediating the induction of NF-κB in neuronal cells, it may be possible to manipulate these diseases for therapeutic gain.
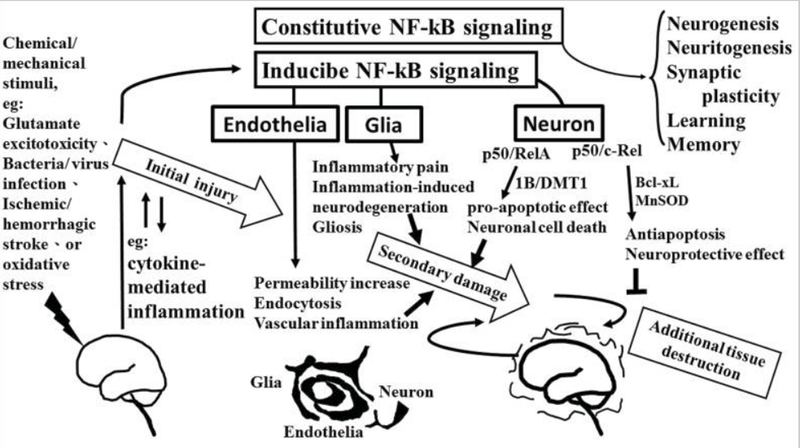
The role of NF-κB in neurological damage. Chemical/mechanical stimulation (such as glutamate excitotoxicity, bacteria/virus infection, ischemic/hemorrhagic stroke, or oxidative stress) to the brain/spinal cord tissue results in initial injury, including glutamate neuron-excitotoxicity, and cytokine-mediated inflammation which increase oxidative stress linking to neuroinflammatory response. NF-κB transcription factors are abundant in the brain where they have diverse functions between neuron, glia, and cerebral blood vessels. Constitutive NF-κB transduction factors are responsible for neurogenesis, neuritogenesis, synaptic plasticity, learning, and memory. Either glial or endothelial inducible NF-κB activation was implicated in neuroinflammation-associated pathogenesis related to secondary neuronal damage, while p50/RelA and p50/c-Rel subunit within activated NF-κB dimers play different roles on neuronal pathogenesis in neuron. The p50/RelA enhances damage by inducing the expression of the 1B isoform of the divalent metal transporter-1(1B/DMT1), p50/c-Rel protects against the damage by increasing the transcription of gene of Bcl-Xl or MnSOD. NF-κB transcription factors have diverse functions that depend on the composition of the NF-κB complex and cell types.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by MOST 104-2320-B-182-010 from Ministry of Science and Technology, Taiwan; EMRPD1E1641 from Ministry of Education, Taiwan; and CMRPD1C0103, CMRPD1B0383, CMRPD1C0563, CMRPD1B0332, and CMRPD1E0341 from Chang Gung Medical Research Foundation, Taiwan.
References
1. Adler A. S., Sinha S., Kawahara T. L., Zhang J. Y., Segal E., Chang H. Y. (2007). Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 21 3244–3257. 10.1101/gad.1588507 [PMC free article] [PubMed] [Cross Ref]
2. Aggarwal B. B. (2004). Nuclear factor-kappaB: the enemy within. Cancer Cell 6 203–208. 10.1016/j.ccr.2004.09.003 [PubMed] [Cross Ref]
3. Ahn H. J., Hernandez C. M., Levenson J. M., Lubin F. D., Liou H. C., Sweatt J. D. (2008). c-Rel, an NF-kappaB family transcription factor, is required for hippocampal long-term synaptic plasticity and memory formation. Learn. Mem. 15 539–549. 10.1101/lm.866408 [PMC free article] [PubMed] [Cross Ref]
4. Ahn K. S., Aggarwal B. B. (2005). Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann. N. Y. Acad. Sci. 1056 218–233. 10.1196/annals.1352.026 [PubMed] [Cross Ref]
5. Akama K. T., Albanese C., Pestell R. G., Van Eldik L. J. (1998). Amyloid beta-peptide stimulates nitric oxide production in astrocytes through an NFkappaB-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 95 5795–5800. 10.1073/pnas.95.10.5795 [PMC free article] [PubMed] [Cross Ref]
6. Akira S., Takeda K. (2004). Functions of toll-like receptors: lessons from KO mice. C. R. Biol. 327 581–589. 10.1016/j.crvi.2004.04.002 [PubMed] [Cross Ref]
7. Anest V., Hanson J. L., Cogswell P. C., Steinbrecher K. A., Strahl B. D., Baldwin A. S. (2003). A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature 423 659–663. 10.1038/nature01648 [PubMed] [Cross Ref]
8. Baeuerle P. A., Baltimore D. (1996). NF-kappa B: ten years after. Cell 87 13–20. 10.1016/S0092-8674(00)81318-5 [PubMed] [Cross Ref]
9. Baeuerle P. A., Henkel T. (1994). Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 12 141–179. 10.1146/annurev.iy.12.040194.001041 [PubMed] [Cross Ref]
10. Baiguera C., Alghisi M., Pinna A., Bellucci A., De Luca M. A., Frau L., et al. (2012). Late-onset Parkinsonism in NFkappaB/c-Rel-deficient mice. Brain 135 2750–2765. 10.1093/brain/aws193 [PMC free article] [PubMed] [Cross Ref]
11. Bakalkin G., Yakovleva T., Terenius L. (1993). NF-kappa B-like factors in the murine brain. Developmentally-regulated and tissue-specific expression. Brain. Res. Mol. Brain Res. 20 137–146. 10.1016/0169-328X(93)90119-A [PubMed] [Cross Ref]
12. Baldwin A. S., Jr. (1996). The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu. Rev. Immunol. 14 649–683. 10.1146/annurev.immunol.14.1.649 [PubMed] [Cross Ref]
13. Barger S. W., Horster D., Furukawa K., Goodman Y., Krieglstein J., Mattson M. P. (1995). Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc. Natl. Acad. Sci. U.S.A. 92 9328–9332. 10.1073/pnas.92.20.9328 [PMC free article] [PubMed] [Cross Ref]
14. Ben-Neriah Y., Karin M. (2011). Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat. Immunol. 12 715–723. 10.1038/ni.2060 [PubMed] [Cross Ref]
15. Bernard D., Quatannens B., Begue A., Vandenbunder B., Abbadie C. (2001). Antiproliferative and antiapoptotic effects of crel may occur within the same cells via the up-regulation of manganese superoxide dismutase. Cancer Res. 61 2656–2664. 10.1016/S0092-8674(00)81318-5 [PubMed] [Cross Ref]
16. Bethea J. R., Castro M., Keane R. W., Lee T. T., Dietrich W. D., Yezierski R. P. (1998). Traumatic spinal cord injury induces nuclear factor-kappaB activation. J. Neurosci. 18 3251–3260. 10.1016/S0092-8674(00)81318-5 [PubMed] [Cross Ref]
17. Bhakar A. L., Tannis L. L., Zeindler C., Russo M. P., Jobin C., Park D. S., et al. (2002). Constitutive nuclear factor-kappa B activity is required for central neuron survival. J. Neurosci. 22 8466–8475. 10.1146/annurev.iy.12.040194.001041 [PubMed] [Cross Ref]
18. Block M. L., Zecca L., Hong J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8 57–69. 10.1038/nrn2038 [PubMed] [Cross Ref]
19. Boissiere F., Hunot S., Faucheux B., Duyckaerts C., Hauw J. J., Agid Y., et al. (1997). Nuclear translocation of NF-kappaB in cholinergic neurons of patients with Alzheimer’s disease. Neuroreport 8 2849–2852. 10.1097/00001756-199709080-00009 [PubMed] [Cross Ref]
20. Brambilla R., Bracchi-Ricard V., Hu W. H., Frydel B., Bramwell A., Karmally S., et al. (2005). Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med. 202 145–156. 10.1084/jem.20041918 [PMC free article] [PubMed] [Cross Ref]
21. Chen C., Edelstein L. C., Gelinas C. (2000). The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol. Cell. Biol. 20 2687–2695. 10.1128/MCB.20.8.2687-2695.2000 [PMC free article] [PubMed] [Cross Ref]
22. Chen L. F., Greene W. C. (2004). Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 5 392–401. 10.1038/nrm1368 [PubMed] [Cross Ref]
23. Crack P. J., Taylor J. M., Ali U., Mansell A., Hertzog P. J. (2006). Potential contribution of NF-kappaB in neuronal cell death in the glutathione peroxidase-1 knockout mouse in response to ischemia-reperfusion injury. Stroke 37 1533–1538. 10.1161/01.STR.0000221708.17159.64 [PubMed] [Cross Ref]
24. DiDonato J. A., Hayakawa M., Rothwarf D. M., Zandi E., Karin M. (1997). A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 388 548–554. 10.1038/41493 [PubMed] [Cross Ref]
25. Duh E. J., Maury W. J., Folks T. M., Fauci A. S., Rabson A. B. (1989). Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc. Natl. Acad. Sci. U.S.A. 86 5974–5978. 10.1073/pnas.86.15.5974 [PMC free article] [PubMed] [Cross Ref]
26. El-Hage N., Bruce-Keller A. J., Yakovleva T., Bazov I., Bakalkin G., Knapp P. E., et al. (2008). Morphine exacerbates HIV-1 Tat-induced cytokine production in astrocytes through convergent effects on [Ca(2+)](i), NF-kappaB trafficking and transcription. PLoS ONE 3 e4093 10.1371/journal.pone.0004093 [PMC free article] [PubMed] [Cross Ref]
27. Farina C., Aloisi F., Meinl E. (2007). Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28 138–145. 10.1016/j.it.2007.01.005 [PubMed] [Cross Ref]
28. Finco T. S., Baldwin A. S. (1995). Mechanistic aspects of NF-kappa B regulation: the emerging role of phosphorylation and proteolysis. Immunity 3 263–272. 10.1016/1074-7613(95)90112-4 [PubMed] [Cross Ref]
29. Ghosh A., Roy A., Liu X., Kordower J. H., Mufson E. J., Hartley D. M., et al. (2007). Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 104 18754–18759. 10.1073/pnas.0704908104 [PMC free article] [PubMed] [Cross Ref]
30. Gray C. M., Remouchamps C., Mccorkell K. A., Solt L. A., Dejardin E., Orange J. S., et al. (2014). Noncanonical NF-kappaB signaling is limited by classical NF-kappaB activity. Sci. Signal. 7 ra13 10.1126/scisignal.2004557 [PMC free article] [PubMed] [Cross Ref]
31. Gupta S. C., Kim J. H., Prasad S., Aggarwal B. B. (2010a). Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev 29 405–434. 10.1007/s10555-010-9235-2 [PMC free article] [PubMed] [Cross Ref]
32. Gupta S. C., Sundaram C., Reuter S., Aggarwal B. B. (2010b). Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 1799 775–787. 10.1016/j.bbagrm.2010.05.004 [PMC free article] [PubMed] [Cross Ref]
33. Hoshino K., Sugiyama T., Matsumoto M., Tanaka T., Saito M., Hemmi H., et al. (2006). IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature 440 949–953. 10.1038/nature04641 [PubMed] [Cross Ref]
34. Hunot S., Brugg B., Ricard D., Michel P. P., Muriel M. P., Ruberg M., et al. (1997). Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 94 7531–7536. 10.1073/pnas.94.14.7531 [PMC free article] [PubMed] [Cross Ref]
35. Ingrassia R., Lanzillotta A., Sarnico I., Benarese M., Blasi F., Borgese L., et al. (2012). 1B/(-)IRE DMT1 expression during brain ischemia contributes to cell death mediated by NF-kappaB/RelA acetylation at Lys310. PLoS ONE 7 e38019 10.1371/journal.pone.0038019 [PMC free article] [PubMed] [Cross Ref]
36. Inta I., Paxian S., Maegele I., Zhang W., Pizzi M., Spano P., et al. (2006). Bim and Noxa are candidates to mediate the deleterious effect of the NF-kappa B subunit RelA in cerebral ischemia. J. Neurosci. 26 12896–12903. 10.1523/JNEUROSCI.3670-06.2006 [PubMed] [Cross Ref]
37. Kaltschmidt B., Kaltschmidt C. (2009). NF-kappaB in the nervous system. Cold Spring Harb. Perspect. Biol. 1 a001271 10.1101/cshperspect.a001271 [PMC free article] [PubMed] [Cross Ref]
38. Kaltschmidt B., Uherek M., Volk B., Baeuerle P. A., Kaltschmidt C. (1997). Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 94 2642–2647. 10.1073/pnas.94.6.2642 [PMC free article] [PubMed] [Cross Ref]
39. Kaltschmidt B., Widera D., Kaltschmidt C. (2005). Signaling via NF-kappaB in the nervous system. Biochim. Biophys. Acta 1745 287–299. 10.1016/j.bbamcr.2005.05.009 [PubMed] [Cross Ref]
40. Kaltschmidt C., Kaltschmidt B., Baeuerle P. A. (1993). Brain synapses contain inducible forms of the transcription factor NF-kappa B. Mech. Dev. 43 135–147. 10.1016/0925-4773(93)90031-R [PubMed] [Cross Ref]
41. Kaltschmidt C., Kaltschmidt B., Baeuerle P. A. (1995). Stimulation of ionotropic glutamate receptors activates transcription factor NF-kappa B in primary neurons. Proc. Natl. Acad. Sci. U.S.A. 92 9618–9622. 10.1073/pnas.92.21.9618 [PMC free article] [PubMed] [Cross Ref]
42. Kaltschmidt C., Kaltschmidt B., Neumann H., Wekerle H., Baeuerle P. A. (1994). Constitutive NF-kappa B activity in neurons. Mol. Cell. Biol. 14 3981–3992. 10.1128/MCB.14.6.3981 [PMC free article] [PubMed] [Cross Ref]
43. Khorooshi R., Babcock A. A., Owens T. (2008). NF-kappaB-driven STAT2 and CCL2 expression in astrocytes in response to brain injury. J. Immunol. 181 7284–7291. 10.4049/jimmunol.181.10.7284 [PubMed] [Cross Ref]
44. Koo J. W., Russo S. J., Ferguson D., Nestler E. J., Duman R. S. (2010). Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl. Acad. Sci. U.S.A. 107 2669–2674. 10.1073/pnas.0910658107 [PMC free article] [PubMed] [Cross Ref]
45. Lanzillotta A., Porrini V., Bellucci A., Benarese M., Branca C., Parrella E., et al. (2015). NF-kappaB in Innate Neuroprotection and Age-Related Neurodegenerative Diseases. Front Neurol 6 98 10.3389/fneur.2015.00098 [PMC free article] [PubMed] [Cross Ref]
46. Lanzillotta A., Sarnico I., Ingrassia R., Boroni F., Branca C., Benarese M., et al. (2010). The acetylation of RelA in Lys310 dictates the NF-kappaB-dependent response in post-ischemic injury. Cell Death Dis 1 e96. 10.1038/cddis.2010.76 [PMC free article] [PubMed] [Cross Ref]
47. Ledoux A. C., Perkins N. D. (2014). NF-kappaB and the cell cycle. Biochem. Soc. Trans. 42 76–81. 10.1042/BST20130156 [PubMed] [Cross Ref]
48. Levenson J. M., Choi S., Lee S. Y., Cao Y. A., Ahn H. J., Worley K. C., et al. (2004). A bioinformatics analysis of memory consolidation reveals involvement of the transcription factor c-rel. J. Neurosci. 24 3933–3943. 10.1523/JNEUROSCI.5646-03.2004 [PubMed] [Cross Ref]
49. Li Q., Verma I. M. (2002). NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2 725–734. 10.1038/nri910 [PubMed] [Cross Ref]
50. Li Z., Nabel G. J. (1997). A new member of the I kappaB protein family, I kappaB epsilon, inhibits RelA (p65)-mediated NF-kappaB transcription. Mol. Cell. Biol. 17 6184–6190. 10.1128/MCB.17.10.6184 [PMC free article] [PubMed] [Cross Ref]
51. Lilienbaum A., Israel A. (2003). From calcium to NF-kappa B signaling pathways in neurons. Mol. Cell. Biol. 23 2680–2698. 10.1128/MCB.23.8.2680-2698.2003 [PMC free article] [PubMed] [Cross Ref]
52. Lin C. C., Hsieh H. L., Shih R. H., Chi P. L., Cheng S. E., Yang C. M. (2013). Up-regulation of COX-2/PGE2 by endothelin-1 via MAPK-dependent NF-kappaB pathway in mouse brain microvascular endothelial cells. Cell Commun Signal 11 8 10.1186/1478-811X-11-8 [PMC free article] [PubMed] [Cross Ref]
53. Lin W. W., Karin M. (2007). A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Invest. 117 1175–1183. 10.1172/JCI31537 [PMC free article] [PubMed] [Cross Ref]
54. Lukiw W. J., Bazan N. G. (1998). Strong nuclear factor-kappaB-DNA binding parallels cyclooxygenase-2 gene transcription in aging and in sporadic Alzheimer’s disease superior temporal lobe neocortex. J. Neurosci. Res. 53 583–592. 10.1002/(SICI)1097-4547(19980901)53:5<583::AID-JNR8>3.0.CO;2-5 [PubMed] [Cross Ref]
55. Mattson M. P. (2005). NF-kappaB in the survival and plasticity of neurons. Neurochem. Res. 30 883–893. 10.1007/s11064-005-6961-x [PubMed] [Cross Ref]
56. Meberg P. J., Kinney W. R., Valcourt E. G., Routtenberg A. (1996). Gene expression of the transcription factor NF-kappa B in hippocampus: regulation by synaptic activity. Brain Res. Mol. Brain Res. 38 179–190. 10.1016/0169-328X(95)00229-L [PubMed] [Cross Ref]
57. Meffert M. K., Baltimore D. (2005). Physiological functions for brain NF-kappaB. Trends Neurosci 28 37–43. 10.1016/j.tins.2004.11.002 [PubMed] [Cross Ref]
58. Meffert M. K., Chang J. M., Wiltgen B. J., Fanselow M. S., Baltimore D. (2003). NF-kappa B functions in synaptic signaling and behavior. Nat. Neurosci. 6 1072–1078. 10.1038/nn1110 [PubMed] [Cross Ref]
59. Mercurio F., Zhu H., Murray B. W., Shevchenko A., Bennett B. L., Li J., et al. (1997). IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278 860–866. 10.1126/science.278.5339.860 [PubMed] [Cross Ref]
60. Niederberger E., Schmidtko A., Gao W., Kuhlein H., Ehnert C., Geisslinger G. (2007). Impaired acute and inflammatory nociception in mice lacking the p50 subunit of NF-kappaB. Eur. J. Pharmacol 559 55–60. 10.1016/j.ejphar.2006.11.074 [PubMed] [Cross Ref]
61. Niranjan R. (2013). Molecular basis of etiological implications in Alzheimer’s disease: focus on neuroinflammation. Mol. Neurobiol. 48 412–428. 10.1007/s12035-013-8428-4 [PubMed] [Cross Ref]
62. Noort A. R., Tak P. P., Tas S. W. (2015). Noncanonical NF-kappaB signaling in rheumatoid arthritis: Dr Jekyll and Mr Hyde? Arthritis Res. Ther. 17:15 10.1186/s13075-015-0527-3 [PMC free article] [PubMed] [Cross Ref]
63. O’Neill L. A., Kaltschmidt C. (1997). NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends in neurosciences 20 252–258. 10.1016/S0166-2236(96)01035-1 [PubMed] [Cross Ref]
64. O’Riordan K. J., Huang I. C., Pizzi M., Spano P., Boroni F., Egli R., et al. (2006). Regulation of nuclear factor kappaB in the hippocampus by group I metabotropic glutamate receptors. J. Neurosci. 26 4870–4879. 10.1523/JNEUROSCI.4527-05.2006 [PubMed] [Cross Ref]
65. Pan W., Yu C., Hsuchou H., Kastin A. J. (2010). The role of cerebral vascular NFkappaB in LPS-induced inflammation: differential regulation of efflux transporter and transporting cytokine receptors. Cell Physiol. Biochem 25 623–630. [PMC free article] [PubMed]
66. Pizzi M., Boroni F., Bianchetti A., Moraitis C., Sarnico I., Benarese M., et al. (2002). Expression of functional NR1/NR2B-type NMDA receptors in neuronally differentiated SK-N-SH human cell line. Eur. J. Neurosci. 16 2342–2350. 10.1046/j.1460-9568.2002.02403.x [PubMed] [Cross Ref]
67. Pizzi M., Sarnico I., Boroni F., Benarese M., Steimberg N., Mazzoleni G., et al. (2005a). NF-kappaB factor c-Rel mediates neuroprotection elicited by mGlu5 receptor agonists against amyloid beta-peptide toxicity. Cell Death. Differ 12 761–772. 10.1038/sj.cdd.4401598 [PubMed] [Cross Ref]
68. Pizzi M., Sarnico I., Boroni F., Benetti A., Benarese M., Spano P. F. (2005b). Inhibition of IkappaBalpha phosphorylation prevents glutamate-induced NF-kappaB activation and neuronal cell death. Acta Neurochir. Suppl. 93 59–63. 10.1007/3-211-27577-0_8 [PubMed] [Cross Ref]
69. Pizzi M., Sarnico I., Lanzillotta A., Battistin L., Spano P. (2009). Post-ischemic brain damage: NF-kappaB dimer heterogeneity as a molecular determinant of neuron vulnerability. FEBS J. 276 27–35. 10.1111/j.1742-4658.2008.06767.x [PubMed] [Cross Ref]
70. Regnier C. H., Song H. Y., Gao X., Goeddel D. V., Cao Z., Rothe M. (1997). Identification and characterization of an IkappaB kinase. Cell 90 373–383. 10.1016/S0092-8674(00)80344-X [PubMed] [Cross Ref]
71. Rolls A., Shechter R., London A., Ziv Y., Ronen A., et al. (2007). Toll-like receptors modulate adult hippocampal neurogenesis. Nat. Cell Biol. 9 1081–1088. 10.1038/ncb1629 [PubMed] [Cross Ref]
72. Sarnico I., Lanzillotta A., Benarese M., Alghisi M., Baiguera C., Battistin L., et al. (2009a). NF-kappaB dimers in the regulation of neuronal survival. Int. Rev. Neurobiol. 85 351–362. 10.1016/S0074-7742(09)85024-1 [PubMed] [Cross Ref]
73. Sarnico I., Lanzillotta A., Boroni F., Benarese M., Alghisi M., Schwaninger M., et al. (2009b). NF-kappaB p50/RelA and c-Rel-containing dimers: opposite regulators of neuron vulnerability to ischaemia. J. Neurochem. 108 475–485. 10.1111/j.1471-4159.2008.05783.x [PubMed] [Cross Ref]
74. Schmidt-Ullrich R., Memet S., Lilienbaum A., Feuillard J., Raphael M., Israel A. (1996). NF-kappaB activity in transgenic mice: developmental regulation and tissue specificity. Development 122 2117–2128. 10.1101/cshperspect.a001271 [PubMed] [Cross Ref]
75. Schmitz M. L., Baeuerle P. A. (1991). The p65 subunit is responsible for the strong transcription activating potential of NF-kappa B. EMBO J. 10 3805–3817. 10.1101/cshperspect.a001271 [PMC free article] [PubMed] [Cross Ref]
76. Schneider A., Martin-Villalba A., Weih F., Vogel J., Wirth T., Schwaninger M. (1999). NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat. Med. 5 554–559. 10.1038/6458 [PubMed] [Cross Ref]
77. Schultz C., Konig H. G., Del Turco D., Politi C., Eckert G. P., Ghebremedhin E., et al. (2006). Coincident enrichment of phosphorylated IkappaBalpha, activated IKK, and phosphorylated p65 in the axon initial segment of neurons. Mol. Cell. Neurosci. 33 68–80. 10.1016/j.mcn.2006.06.008 [PubMed] [Cross Ref]
78. Sen R., Baltimore D. (1986). Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell 47 921–928. 10.1016/0092-8674(86)90807-X [PubMed] [Cross Ref]
79. Shih R. H., Yang C. M. (2010). Induction of heme oxygenase-1 attenuates lipopolysaccharide-induced cyclooxygenase-2 expression in mouse brain endothelial cells. J Neuroinflammation 7 86 10.1186/1742-2094-7-86 [PMC free article] [PubMed] [Cross Ref]
80. Stone K. P., Kastin A. J., Pan W. (2011). NFkB is an unexpected major mediator of interleukin-15 signaling in cerebral endothelia. Cell Physiol. Biochem 28 115–124. 10.1159/000331720 [PMC free article] [PubMed] [Cross Ref]
81. Sun S. C. (2012). The noncanonical NF-kappaB pathway. Immunol. Rev. 246 125–140. 10.1111/j.1600-065X.2011.01088.x [PMC free article] [PubMed] [Cross Ref]
82. Tak P. P., Gerlag D. M., Aupperle K. R., Van De Geest D. A., Overbeek M., Bennett B. L., et al. (2001). Inhibitor of nuclear factor kappaB kinase beta is a key regulator of synovial inflammation. Arthritis Rheum. 44 1897–1907. 10.1002/1529-0131(200108)44:8<1897::AID-ART328>3.0.CO;2-4 [PubMed] [Cross Ref]
83. Thanos D., Maniatis T. (1995). NF-kappa B: a lesson in family values. Cell 80 529–532. 10.1016/0092-8674(95)90506-5 [PubMed] [Cross Ref]
84. Tilstra J. S., Robinson A. R., Wang J., Gregg S. Q., Clauson C. L., Reay D. P., et al. (2012). NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Invest. 122 2601–2612. 10.1172/JCI45785 [PMC free article] [PubMed] [Cross Ref]
85. Tsai C. L., Chen W. C., Hsieh H. L., Chi P. L., Hsiao L. D., Yang C. M. (2014). TNF-alpha induces matrix metalloproteinase-9-dependent soluble intercellular adhesion molecule-1 release via TRAF2-mediated MAPKs and NF-kappaB activation in osteoblast-like MC3T3-E1 cells. J. Biomed. Sci. 21 12 10.1186/1423-0127-21-12 [PMC free article] [PubMed] [Cross Ref]
86. Valerio A., Boroni F., Benarese M., Sarnico I., Ghisi V., Bresciani L. G., et al. (2006). NF-kappaB pathway: a target for preventing beta-amyloid (Abeta)-induced neuronal damage and Abeta42 production. Eur. J. Neurosci. 23 1711–1720. 10.1111/j.1460-9568.2006.04722.x [PubMed] [Cross Ref]
87. Valerio A., Dossena M., Bertolotti P., Boroni F., Sarnico I., Faraco G., et al. (2009). Leptin is induced in the ischemic cerebral cortex and exerts neuroprotection through NF-kappaB/c-Rel-dependent transcription. Stroke 40 610–617. 10.1161/STROKEAHA.108.528588 [PubMed] [Cross Ref]
88. Vallabhapurapu S., Karin M. (2009). Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 27 693–733. 10.1146/annurev.immunol.021908.132641 [PubMed] [Cross Ref]
89. Verma I. M., Stevenson J. K., Schwarz E. M., Van Antwerp D., Miyamoto S. (1995). Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 9 2723–2735. 10.1101/gad.9.22.2723 [PubMed] [Cross Ref]
90. Wellmann H., Kaltschmidt B., Kaltschmidt C. (2001). Retrograde transport of transcription factor NF-kappa B in living neurons. J. Biol. Chem. 276 11821–11829. 10.1074/jbc.M009253200 [PubMed] [Cross Ref]
91. Whiteside S. T., Epinat J. C., Rice N. R., Israel A. (1997). I kappa B epsilon, a novel member of the I kappa B family, controls RelA and cRel NF-kappa B activity. EMBO J. 16 1413–1426. 10.1093/emboj/16.6.1413 [PMC free article] [PubMed] [Cross Ref]
92. Yakovleva T., Bazov I., Watanabe H., Hauser K. F., Bakalkin G. (2011). Transcriptional control of maladaptive and protective responses in alcoholics: a role of the NF-kappaB system. Brain Behav. Immun. 25(Suppl. 1), S29–S38. 10.1016/j.bbi.2010.12.019 [PMC free article] [PubMed] [Cross Ref]
93. Yamamoto Y., Verma U. N., Prajapati S., Kwak Y. T., Gaynor R. B. (2003). Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature 423 655–659. 10.1038/nature01576 [PubMed] [Cross Ref]
94. Yang C. M., Chien C. S., Hsiao L. D., Luo S. F., Wang C. C. (2002). Interleukin-1beta-induced cyclooxygenase-2 expression is mediated through activation of p42/44 and p38 MAPKS, and NF-kappaB pathways in canine tracheal smooth muscle cells. Cell. Signal. 14 899–911. 10.1016/S0898-6568(02)00037-2 [PubMed] [Cross Ref]
95. Yang C. M., Luo S. F., Hsieh H. L., Chi P. L., Lin C. C., Wu C. C., et al. (2010). Interleukin-1beta induces ICAM-1 expression enhancing leukocyte adhesion in human rheumatoid arthritis synovial fibroblasts: involvement of ERK, JNK, AP-1, and NF-kappaB. J. Cell. Physiol. 224 516–526. 10.1002/jcp.22153 [PubMed] [Cross Ref]
96. Zandi E., Rothwarf D. M., Delhase M., Hayakawa M., Karin M. (1997). The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 91 243–252. 10.1016/S0092-8674(00)80406-7 [PubMed] [Cross Ref]
97. Zhang G., Li J., Purkayastha S., Tang Y., Zhang H., Yin Y., et al. (2013). Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature 497 211–216. 10.1038/nature12143 [PMC free article] [PubMed] [Cross Ref]
98. Zhang H., Hilton M. J., Anolik J. H., Welle S. L., Zhao C., Yao Z., et al. (2014). NOTCH inhibits osteoblast formation in inflammatory arthritis via noncanonical NF-kappaB. J. Clin. Invest. 124 3200–3214. 10.1038/nri910 [PMC free article] [PubMed] [Cross Ref]
___________________________________________________________________________________________________________________________
NF-kB Transcription Factors
Dr. Thomas D. Gilmore
Biology Department, Boston University
5 Cummington Mall,
Boston, Massachusetts 02215-2406, USA
(617) 353-5444 or 5445; fax (617) 353-6340
The Rel/NF-kappaB Signal Transduction Pathway
Rel or NF-kappaB (NF-kB) proteins comprise a family of structurally-related eukaryotic transcription factors that are involved in the control of a large number of normal cellular and organismal processes, such as immune and inflammatory responses, developmental processes, cellular growth, and apoptosis. In addition, these transcription factors are persistently active in a number of disease states, including cancer, arthritis, chronic inflammation, asthma, neurodegenerative diseases, and heart disease (see DISEASES link).
Rel/NF-kB transcription factors include a collection of proteins, with functions conserved from the fruit fly Drosophila melanogaster to humans; more recently, Rel/NF-kB homologs have also been found to occur even in organisms as simple as Cnidarians (e.g., sea anemones and corals), Porifera (sponges) and the single-celled eukaryote Capsaspora owczarzaki, Among the commonly studied model organisms, these transcription factors are notably absent in yeast and the nematode Caenorhabditis elegans (the latter having apparently lost the pathway during evolution); in part, the absence of the NF-kB pathway in yeast may be because one of the primary roles of these factors is to control a variety of physiological aspects of immune and inflammatory responses.
Rel/NF-kB proteins are related through a highly conserved DNA-binding/dimerization domain called the Rel homology (RH) domain. However, Rel/NF-kB proteins can be divided into two classes based on sequences C-terminal to the RH domain (Figure 1). Members of one class (the NF-kB proteins p105, p100, and Drosophila Relish) have long C-terminal domains that contain multiple copies of ankyrin repeats, which act to inhibit these molecules. Members of the NF-kB class become active, shorter DNA-binding proteins (p105 to p50, p100 to p52) by either limited proteolysis or arrested translation. As such, members of this first class are generally not activators of transcription, except when they form dimers with members of the second class of Rel/NF-kB transcription factors. The second class (the Rel proteins) includes c-Rel (and its retroviral homologue v-Rel), RelB, RelA (p65), and the Drosophila Dorsal and Dif proteins. This second class of Rel proteins contains C-terminal transcription activation domains, which are often not conserved at the sequence level across species, even though they can activate transcription in a variety of species. The cDNA and predicted protein sequences of Rel/NF-kB transcription factors can be rapidly accessed via this site (see SEQUENCES link).
Rel/NF-kB transcription factors bind to 9-10 base pair DNA sites (called kB sites) as dimers. All vertebrate Rel proteins can form homodimers or heterodimers, except for RelB, which can only form heterodimers. This combinatorial diversity contributes to the regulation of distinct, but overlapping, sets of genes, in that the individual dimers have distinct DNA-binding site specificities for a collection of related kB sites. The term NF-kappaB commonly refers specifically to a p50-RelA heterodimer, which is one of the most avidly forming dimers and is the major Rel/NF-kB complex in most cells. The x-ray crystallographic structures of several Rel/NF-kB dimers on DNA (including p50-p50, p65-p65, p50-p65, c-Rel-c-Rel, p50-p65-IkB) have now been solved, and these structures can be accessed from this site (see STRUCTURES link)
The activity of NF-kB is primarily regulated by interaction with inhibitory IkB proteins. As with the Rel/NF-kB proteins, there are several IkB proteins, which have different affinities for individual Rel/NF-kB complexes, are regulated slightly differently, and are expressed in a tissue-specific manner. The IkB proteins include, at least, p105, p100, IkBa, IkBb, IkBg, IkBe, IkBz, Bcl-3, and the Drosophila Cactus protein. The cDNA and predicted protein sequences of these IkBs can be obtained through this site (see SEQUENCES link).
The best-studied NF-kB-IkB interaction is that of IkBa with the NF-kB p50-RelA dimer. This interaction blocks the ability of NF-kB to bind to DNA and results in the NF-kB complex being primarily in the cytoplasm due to a strong nuclear export signal in IkBa. That is, the NF-kB-IkBa complex is continuously shuttling between the nucleus and the cytoplasm, but its rate of nuclear export exceeds its rate of import and thus the complex is generally cytoplasmic. From biochemical studies and direct structural determinations (see links at this site), it is clear that IkBa makes multiple contacts with NF-kB. These interactions cover sequences of NF-kB that are important for DNA binding. In contrast, when IkBb interacts with the NF-kB complex, the complex is retained in the cytoplasm (i.e., does not undergo nucleo-cytoplasmic shuttling). Thus, not all NF-kB-IkB interactions are the same.
In most cells, NF-kB is present as a latent, inactive, IkB-bound complex in the cytoplasm. When a cell receives any of a multitude of extracellular signals (see INDUCERS link), NF-kB rapidly enters the nucleus and activates gene expression (see TARGET GENES link). Therefore, a key step for controlling NF-kB activity is the regulation of the IkB-NF-kB interaction. Many of the molecular details of this control are now understood (Figure 2). Almost all signals that lead to activation of NF-kB converge on the activation of a high molecular weight complex that contains a serine-specific IkB kinase (IKK). IKK is an unusual kinase in that in most cells IKK contains (at least) three distinct subunits: IKKalpha, IKKbeta and IKKgamma. IKKa and IKKb are related catalytic kinase subunits, and IKKg (aka NEMO) is a regulatory subunit that serves as a sensing scaffold and integrator of upstream signals for activation of the catalytic subunits. In the classical or canonical pathway, activation of IKK complex leads to the phosphorylation by IKKb of two specific serines near the N terminus of IkBa, which targets IkBa for ubiquitination (generally by a complex called beta-TrCP) and degradation by the 26S proteasome. In the non-canonical (or alternative) pathway, the p100-RelB complex is activated by phosphorylation of the C-terminal region of p100 by an IKKa homodimer (lacking IKKgamma), which leads to ubiquitination followed by degradation of the p100 IkB-like C-terminal sequences to generate p52-RelB. In either pathway, the unmasked NF-kB complex can then enter the nucleus to activate target gene expression. In the classical pathway, one of the target genes activated by NF-kB is that which encodes IkBa. Newly-synthesized IkBa can enter the nucleus, remove NF-kB from DNA, and export the complex back to the cytoplasm to restore the original latent state. Thus, the activation of the NF-kB pathway is generally a transient process, lasting from 30-60 minutes in most cells.
A variety of recent evidence, however, indicates that the control of the NF-kB pathway is more complex than simply IKK-mediated regulation of the IkB-NF-kB interaction. For example, RelA and p50 are regulated by ubiquitination, acetylation, methylation, phosphorylation, oxidation/reduction, and prolyl isomerization. Moreover, as a consequence of induction of NF-kB activity (at least by tumor necrosis factor) IKKa is also induced to enter the nucleus where it becomes associated with kB site promoters/enhancers to phosphorylate histone H3 which enhances the transcription of kB site-dependent genes. Finally, proteins in the NF-kB signaling pathway participate in a number of protein-protein interactions with non-NF-kB proteins (see PROTEIN-PROTEIN INTERACTIONS link).
In some normal cells, such as B cells, some T cells, Sertoli cells and some neurons, NF-kB is constitutively located in the nucleus. In addition, in many cancer cells (including breast cancer, colon cancer, prostate cancer, lymphoid cancers, and probably many others; see DISEASES link) NF-kB is constitutively active and located in the nucleus. In some cancers, this is due to chronic stimulation of the IKK pathway, while in other cases (such as some Hodgkin’s and diffuse large B-cell lymphoma cells) the gene encoding IkB can be mutated and defective. Moreover, several human lymphoid cancer cells have mutations or amplifications of genes encoding Rel/NF-kB transcription factors (esp REL in human B-cell lymhoma) and many multiple myelomas have mutations in genes encoding NF-kB signaling regulatory proteins that lead to constitutive activation of NF-kB. It is thought that continuous nuclear Rel/NF-kB activity protects cancer cells from apoptosis and in some cases stimulates their growth. Therefore, many current anti-tumor therapies seek to block NF-kB activity as a means to inhibit tumor growth or to sensitize the tumor cells to more conventional therapies, such as chemotherapy.
The Rel/NF-kB family is arguably the most-studied collection of eukaryotic transcription factors. For a collection of reviews on these transcription factors, the reader is directed to the November 22, 1999 and October 30, 2006 issues of Oncogene, which contain a series of reviews on Rel/NF-kB.
Our extensive knowledge of Rel/NF-kB signaling exposes also the reaches of our ignorance. We still have very little understanding of the complex in vivo dynamics of this pathway. For example, in most cell types and signaling conditions, it is still not known what are the contributions of specific Rel/NF-kB complexes (p50-RelA vs. p52-c-Rel vs. c-Rel-c-Rel) to most physiological responses. Over-expression studies in tissue culture almost certainly do not accurately reflect physiological signaling events. Similarly, what controls the balance between the levels of the various heterodimeric complexes in vivo is not known. Studies in Drosophila have elegantly shown that very small differences in nuclear concentrations of these factors, in their affinities for target DNA sites, and in cooperation or competition between Rel proteins and other transcription factors can have profound physiological consequences in organisms. Lastly, in many situations, it is not known how or which of the many genes induced by Rel/NF-kB factors in a given response contribute to that response. The development of methods to analyze genome-wide changes in gene expression (e.g., cDNA microarrays), which has already begun to uncover additional Rel/NF-kB-responsive genes, has helped to clarify which Rel/NF-kB target genes are activated in a given response.
As described above, the structures of several Rel/NF-kB dimers on DNA or bound to IkB are known. In all cases, these structures have been derived from molecules that contain almost exclusively residues from the RH domain. As such, these studies provide rather static glimpses of these factors at work. Several molecular and biochemical studies indicate that Rel dimers assume distinct conformations when bound to DNA versus as free or IkB-bound dimers or when bound to different kB sites. Moreover, such studies have also indicated that C-terminal residues influence sequences within the RH domain. Furthermore, there is surprisingly little information about how any of the Rel/NF-kB complexes actually activate transcription when bound to DNA: that is, what are the co-activators or basal factors with which they interact to activate transcription? Therefore, we cannot accurately simulate the dynamic nature of the complex as it releases from IkB, enters the nucleus, binds to DNA, and enhances gene expression; however, mathematical and computational modeling of the NF-kB pathway is beginning to address the dynamics of the pathway in response to various signals. In addition, recent studies suggest that NF-kB complexes bind to specific promoters in a dynamic on-off manner, with occupancy of a specific promoter sequence by an individual NF-kB dimer lasting on the order of seconds.
Although the discovery and characterization of the IkB kinase complex was a monumental step in our understanding of the regulation of this pathway, it raised almost as many questions as it has answered. For example, the following issues remain murky: 1) precisely which proteins are in the IKK complex in all cell types; 2) the exact size of the complex in all cell types; 3) what is the physiological relevance of phosphorylation by the IKK complex of substrates other than IkB; 4) how the various NF-kB and non-NF-kB activation pathways converge on IKK (for example, what and how many upstream kinases can activate IKK); 5) how is IKK activated by what appears to be induced clustering; 6) how is it that one subunit of this complex (IKKa) controls a specific developmental process, namely keratinocyte differentiation; 7) all of the other signaling pathways that crosstalk via or to IKK; 8) how do the two catalytic kinases within the IKK complex act on substrate proteins; and 9) how do reactive oxygen species and reactive Cysteine residues impact IKK activity. Recent X-ray crystal structural information on the IKK protein and components of the IKK complex may help answer some of these questions.
The study of v-Rel has unequivocally demonstrated that Rel/NF-kB transcription factors can be oncogenic, and one would like to know how the activating mutations in v-Rel have altered its structure as compared to c-Rel. However, v-Rel has accumulated so many activating mutations that it may not be a precise model for the role of these transcription factors in human cancers, where a single mutation (or gene amplification event) has occurred. Thus, in some cases, it is not known whether the rearrangements, mutations, and amplifications in Rel/NF-kB/IkB genes that have been repeatedly identified in several human cancers and the constitutive NF-kB signaling seen in certain human cancers or induced by oncogenic human viruses (e.g., EBV and HTLV-1) contribute to proliferation, abrogate growth suppression, influence the control of apoptosis, or affect all of these processes.
The extensive involvement of Rel/NF-kB transcription factors in human inflammation and disease establishes them as targets for therapeutics. Indeed, many common synthetic (e.g., aspirin), and traditional (e.g., green tea, curcumin) remedies target, at least in part, the Rel/NF-kB signaling pathway. However, there are over 800 compounds that have been shown to inhibit NF-kB signaling (see INHIBITORS at this site), and thus, the physiological or pharmacological utility of using any single compound for inhibition of NF-kB activity is a bit muddled. Nevertheless, our knowledge of the molecular details of this pathway is enabling the development of more specific and potent inhibitors of NF-kB signaling, and indeed, some NF-kB signaling inhibitors are entering clinical trials.
Among the many publications on this topic, there are inconsistencies in the naming of genes and proteins in the Rel/NF-kB pathway. Although a system of nomenclature for the Rel/NF-kB transcription factors and IkB proteins was established previously (Nabel and Verma, 1993), we use and suggest a slightly modified nomenclature (Table 1). The revised nomenclature reflects the new members of this pathway, common usage over the past several years, and at times my own judgment. In most cases, the choice was quite simple, although the p65 vs. RelA decision continues to be a thorny one.
Research in the Gilmore laboratory has been supported by the National Cancer Institute of the National Institutes of Health, the National Science Foundation, the American Cancer Society, the Cure for Lymphoma Foundation, the Leukemia Research Foundation, the Council for Tobacco Research, and Boston University. For more information on the Gilmore lab, go to the link for THE LAB.
Beyaert R (editor) (2004). Nuclear Factor-kappaB: Regulation and Role in Disease. Kluwer Academic Publishes, Dordrecht, The Netherlands. 426 pages
Gilmore TD (editor) (2006). NF-kB: from basic research to human disease. Oncogene (Reviews) 51: 6679-6899
Nabel GJ and Verma IM.(1993). Genes & Development 7: 2063
Perkins ND (2007) Integrating cell-signaling pathways with NF-kB and IKK function. Nature Reviews Molecular Cell Biology 8: 40-62
Figure 1 Structures of Rel family transcription factors. Shown are the generalized structures of the two classes of Rel transcription factors. All have a conserved DNA-binding/dimerization domain called the Rel homology (RH) domain, which also has sequences important for nuclear localization (N) and IkB inhibitor binding. Class I proteins have additional inserted sequences in the RH domain. The C-terminal halves of the class I Rel proteins have ankyrin repeat-containing inhibitory domains, which can be removed by proteasome-mediated proteolysis (PROTEASE). The C-terminal halves of the class II Rel proteins have transcriptional activation domains.
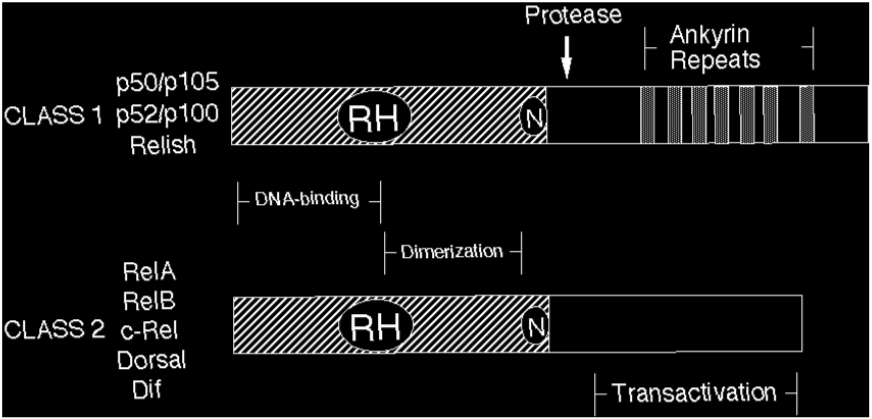
Figure 2 Rel/NF-kB signal transduction. In the classical pathway, various signals converge on activation of the IkB kinase (IKK) complex. IKK then phosphorylates IkB at 2 N-terminal serines, which signals it for ubiquitination and proteolysis. Freed NF-kB (p50-RelA, in this case) enters the nucleus and activates gene expression. One NF-kB target gene encodes IkB. The newly synthesized IkB can enter the nucleus, pull NF-kB off DNA, and export NF-kB back to its resting state in the cytoplasm. Thick lines indicate the activating pathway; thin lines indicate the inactivating pathway.
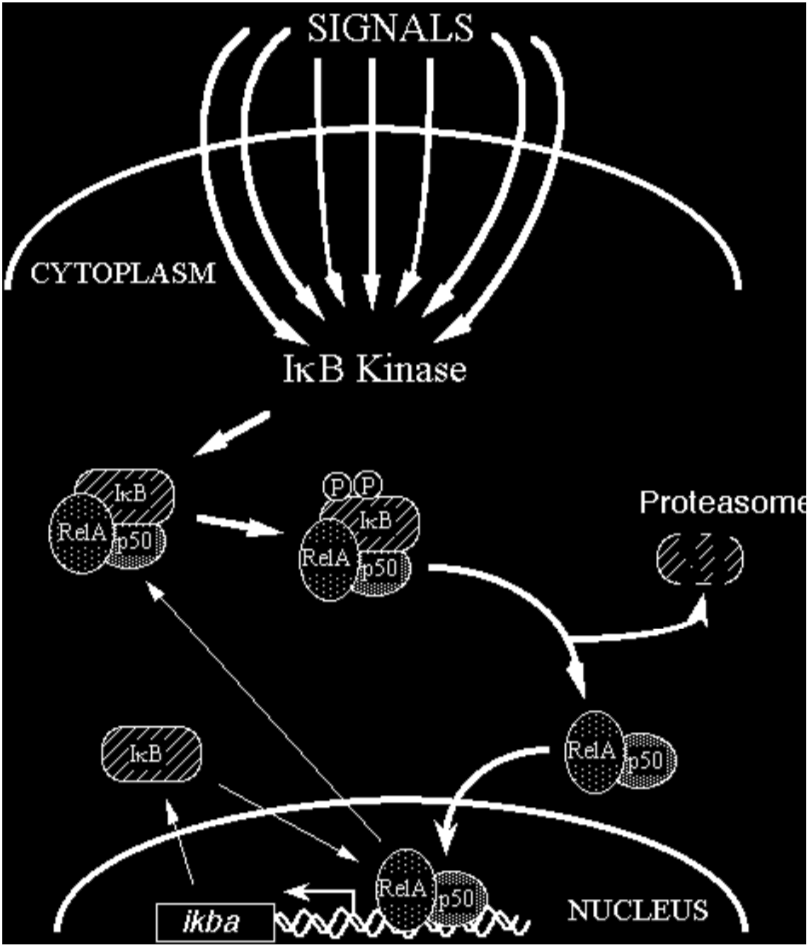
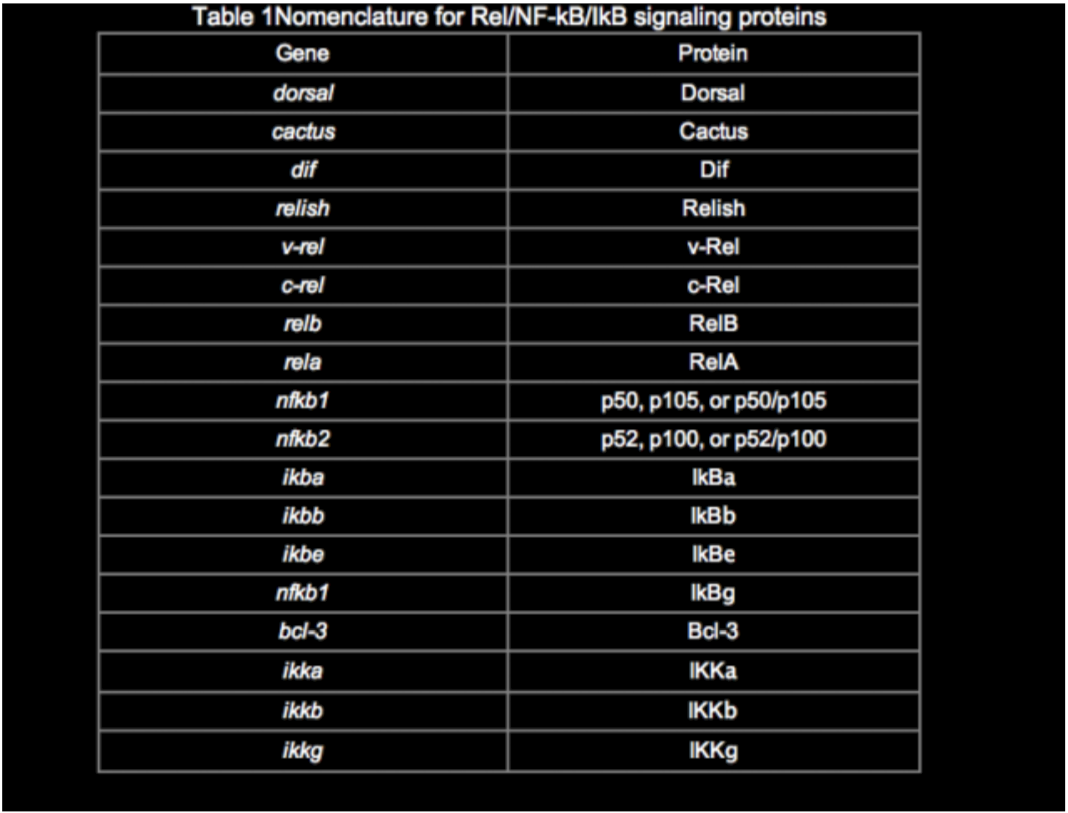
_____________________________________________________________________________________________________________________
Anticonvulsants (also commonly known as antiepileptic drugs or as antiseizure drugs) are a diverse group of pharmacological agents used in the treatment of epileptic seizures. Anticonvulsants are also increasingly being used in the treatment of bipolar disorder [1] and borderline personality disorder, [2] since many seem to act as mood stabilizers, and for the treatment of neuropathic pain. [3] Anticonvulsants suppress the excessive rapid firing of neurons during seizures.[4] Anticonvulsants also prevent the spread of the seizure within the brain.[5]
Conventional antiepileptic drugs may block sodium channels or enhance γ-aminobutyric acid (GABA) function. Several antiepileptic drugs have multiple or uncertain mechanisms of action.[6] Next to the voltage-gated sodium channels and components of the GABA system, their targets include GABAA receptors, the GAT-1 GABA transporter, and GABA transaminase. [7] Additional targets include voltage-gated calcium channels, SV2A, and α2δ.[8][9] By blocking sodium or calcium channels, antiepileptic drugs reduce the release of excitatory glutamate, whose release is considered to be elevated in epilepsy, but also that of GABA.[10] This is probably a side effect or even the actual mechanism of action for some antiepileptic drugs, since GABA can itself, directly or indirectly, act proconvulsively.[10]
Another potential target of antiepileptic drugs is the peroxisome proliferator-activated receptor alpha.[11][12][13][14][15][16][17] The drug class was the 5th-best-selling in the US in 2007.[18]
Some anticonvulsants have shown antiepileptogenic effects in animal models of epilepsy.[19] That is, they either prevent the development of epilepsy or can halt or reverse the progression of epilepsy. However, no drug has been shown in human trials to prevent epileptogenesis (the development of epilepsy in an individual at risk, such as after a head injury).
It was concluded form the study that COX-2 mediated anti-proliferation was responsible for the cytotoxic activity of withanolide sulfoxide (83) (Mulabagal et al., 2009). Moreover, gingerol (84) (Figure 8) is polyphenolic compound, which can be found in majority of Zingiberaceae species. A study was designed to evaluate the effect of gingerol on streptozotocin induced Alzheimer diseases in vivo by using whiskers rat model. The basic purpose of the study was to determine that whether the treatment with gingerol can to reduce the inflammation and improve the cognitive impairment in sporadic Alzheimer's disease in whiskers rat model. The results suggested that pretreatment with 10 and 20 mg/kg bwt significantly reduce the α-, β- secretases, APH1a and COX-2 levels, which resulted in the improvement of cognitive behaviors (El Halawany et al., 2017). Likewise similar activity was reported for two styryllactone, altholactone (85) and (+)-goniothalmin (86) (Figure 8) isolated from Alphonsea javanica. It was concluded form the current study that both compounds significantly reduced the LPS-induced NO production, phosphorylation of IKB alpha, and also down regulated the gene expression of iNOS and COX-2 (Johnson et al., 2013). Moreover, diglyceride (87) from Amaranthus tricolor were also able to inhibit the COX-1 enzyme by 78, 63, and 93%, respectively and the COX-2 enzyme by 87, 74, and 95%, respectively (Jayaprakasam et al., 2004). However, due to their non-selective COX activity, it is expected that use of these diglyceride (87) might be associated with undesirable gastrointestinal effects, which limits its use as therapeutic application. Ligustilide (88) (Figure 8), phthalide derivative from Angelica tenuissi was also subjected to anti-inflammatory evaluation. The bioassay results demonstrated that Ligustilide (87) has potent anti-inflammatory activity by modulating NF-κB and MAPK pathways, which led to the suppression of LPS-induced iNOS and COX-2 expression at both mRNA and protein levels. Furthermore, Ligustilide (88) additionally inhibited the phosphorylation of IkBa and p38 mitogen-activated protein kinase in a dose dependent manner (Chung et al., 2012). Bruceine D (89) (Figure 8), belong to unique class of compound, which is mostly predominant in Simaroubaceae family. Till this date the pharmacological profile on this class is yet to be established. However, in recent study bruceine D showed significant inhibitory activity against COX-2 with an IC50 value of 20.6 μM demonstrating the anti-inflammatory potential of this unique class (Su et al., 2002). Likewise corresponding activity was observed in a study conducted on handelin (90) (Figure 8), duaianolide dimer isolated from Chrysanthemum boreale. It was observed that handelin (90) suppressed NO and PGE2 activity via suppressing the iNOS and COX-2 expression in LPS-induced RAW 264.7 macrophage cells. In addition, pretreatment with handelin (90) also deceased the production of IL-1β and TNF-α in a dose dependently manner. Further mechanist study revealed that handelin (90) also inhibited the activation and translocation of NF-κB into the nucleus, which is in complete conjunction with the suppression of COX-2 and iNOS activity.
Structure of miscellaneous with potent COX-2 and PGE2 inhibitory activity (80) taiwanin C (81), balanophonin (82), sitoindoside IX (83), withanolide sulfoxide (84), gingcrol, (85) altholactone (86), goniothalmin (87), diglyccride (88), ligustilide (89), bruceine D (90), bandelin (91), guggulstcrone (92), isoliquiritin (93), isoliquiritigenin (94), sphondin (95), 4-Methoxyhonokol (96), Schisandrin (97), bocravinone B.
Reference for ATP
Robitaille R.
Purinergic receptors and their activation by endogenous purines at perisynaptic glial cells of the frog neuromuscular junction. J. Neurosci. 1995; 15: 7121-7131.
PubMed
Stevens B. Fields R.D. Response of Schwann cells to action potentials in development. Science. 2000; 287: 2267-2271.
Scopus (196)
Araque A. et al.
Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999; 22: 208-215.
Scopus (1247)
Google Scholar
LevRam V. , Ellisman M.H.
Axonal activation-induced calcium transients in myelinating Schwann cells, sources and mechanisms. J. Neurosci. 1995; 15: 2628-2637
PubMed
Bergles D.E. et al. Glutamatergic synapses on oligo-dendrocyte precursor cells in the hippocampus. Nature. 2000; 405: 187-191.
Scopus (531)
PubMed
Araque A. et al. SNARE protein-dependent glutamate release from astrocytes. J. Neurosci. 2000; 15: 666-673.
Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994; 263: 1768-1771.
Scopus (760)
PubMed
Parpura V. et al. Glutamate-mediated astrocyte–neuron signaling. Nature. 1994; 369: 744-747.
Scopus (1213)
PubMed
Demerens C. et al. Induction of myelination in the central nervous system by electrical activity.
Proc. Natl. Acad. Sci. U. S. A. 1996; 93: 9887-9892.
Scopus (398)
PubMed
Stevens B. et al. Control of myelination by specific patterns of neural impulses. J. Neurosci. 1998; 18: 9303-9311.
Son Y.J. et al. Schwann cells induce and guide sprouting and reinnervation of neuromuscular junctions. Trends Neurosci. 1996; 19: 280-285.
Scopus (165)
PubMed
Jahromi B.S. et al. Transmitter release increases intracellular calcium in perisynaptic Schwann cells in situ. Neuron. 1992; 8: 1069-1077.
Scopus (163)
PubMed
Reist N.E., Smith S.J. Neurally evoked calcium transients in terminal Schwann cells at the neuromuscular junction. Proc. Natl. Acad. Sci. U. S. A. 1992; 89: 7625-7629.
Scopus (92)
PubMed
Georgiou J. et al. Synaptic regulation of glial protein expression in vivo. Neuron. 1994; 12: 443-455.
Scopus (83)
PubMed
Google Scholar, Robitaille R. Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron. 1998; 21: 847-855.
Scopus (185)
PubMed
Parpura V. et al. Neuroligand-evoked calcium-dependent release of excitatory amino acids from Schwann cells. J. Neurosci. 1995; 15: 5831-5839.
Ralevic V. ,Burnstock G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998; 50: 413-492
Dubyak G.R. , El-Moatassim C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am. J. Physiol. 1993; 265: C577-C606.
Gousley G.D. Physiological effects of extracellular nucleotides in the inner ear. Clin. Exp. Pharmacol. Physiol. 2000; 27: 575-580.
Osipchuk Y. Cahalan M. Cell-to-cell spread of calcium signals mediated by ATP receptors in mast cells. Nature. 1992; 359: 241-244.
Scopus (304)
PubMed
Guthrie P.B. et al. ATP release from astrocytes mediates glial calcium waves. J. Neurosci. 1999; 19: 520-528.
Cornell-Bell A.H. et al. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990; 247: 470-473.
Scopus (1209)
PubMed
Fam S.R. et al. P2Y1 purinoceptor-mediated Ca2+ signaling and Ca2+ wave propogation in dorsal spinal astrocytes. J. Neurosci. 2000; 20: 2800-2808.
Newman E.A. Zahs K.R. Calcium waves in retinal glial cells. Science. 1997; 275: 844-847.
Scopus (326)
PubMed
Newman E.A. Zahs K.R. Modulation of neuronal activity by glial cells in the retina. J. Neurosci. 1998; 18: 4022-4028.
Jeftinija S.D. Jeftinija K.V. ATP stimulates release of excitatory amino acids from cultured Schwann cells. Neuroscience. 1998; 82: 927-934.
Scopus (48)
PubMed
Edwards F.A. et al. ATP receptor-mediated synaptic currents in the central nervous system. Nature. 1992; 359: 144-147.
Scopus (679)
PubMed
Gu J.G., MacDermott A.B. Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature. 1997; 389: 749-753.
Scopus (384)
PubMed
Zimmermann H. Signaling via ATP in the nervous system. Trends Neurosci. 1994; 17: 420-426.
Scopus (399)
PubMed
Jo Y-H., Schlichter R. Synaptic corelease of ATP and GABA in cultured spinal neurons.
Nat. Neurosci. 1999; 2: 241-245.
Scopus (266)
PubMed
Bo X. Brunstock G. Distribution of 3 Araque A. et al. Tripartite synapses: glia, the unacknowledged partner.Trends Neurosci.1999; 22: 208-215.
Scopus (1247)
H] a, beta-methylene ATP binding sites in rat brain and spinal cord. NeuroReport. 1994; 5: 1601-1604.
Scopus (86)
Von Kugelgen I. et al. Role of action potentials and calcium influx in ATP- and UDP- induced noradrenaline release from rat cultured sympathetic neurones. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999; 359: 360-369.
Scopus (34)
PubMed
Silinsky E.M. et al. Quantal ATP release from motor nerve endings and its role in neurally mediated depression. Prog. Brain Res. 1999; 120: 145-158.
Zimmermann H. New insights into molecular structure and function of ectonucleosidases in the nervous system. Neurochem. Int. 1998; 32: 421-425.
Scopus (116)
PubMed
Santos P.F. et al. Characterization of ATP release from cultures enriched in cholinergic amacrine-like neurons. J. Neurobiol. 1999; 41: 340-348.
Scopus (65)
PubMed
Inoue K.I. The functions of ATP receptors in the hippocampus. Pharmacol. Res. 1998; 38: 323-331.
Scopus (31)
PubMed
Wachtler J. et al. Activity-dependent intracellular calcium transients in unmyelinated nerve fibers of the isolated adult rat vagus nerve. Pflügers Arch. Eur. J. Physiol. 1998; 435: 678- 686.
Scopus (38)
PubMed
Kriegler S. Chiu S.Y. Calcium signaling of glial cells along mammalian axons. J. Neurosci. 1993; 13: 4229-4245.
Kuperman A.S. et al. Release of adenine nucleotides from nerve axons. Nature. 1964; 204: 1000-1001.
Scopus (19)
PubMed
Weinneich D. Hammerschlag R. Nerve impulse-enhanced release of amino acids from non- synaptic regions of peripheral and central nerve trunks of bullfrog. Brain Res. 1975; 84: 137-142.
Scopus (78)
PubMed
Reisin I.L. et al. The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel. J. Biol. Chem. 1994; 269: 20584-20591.
Abraham E.H. et al. The multidrug resistance (mdr1) gene product functions as an ATP channel. Proc. Natl. Acad. Sci. U. S. A. 1993; 90: 312-316.
Scopus (336)
PubMed
Reddy M.M. et al. Failure of the cystic fibrosis transmembrane conductance regulator to conduct ATP. Science. 1996; 271: 1876-1879.
Scopus (150)
PubMed
Grygorczyk R. Hanrahan J.W. CFTR-dependent ATP release from epithelial cells triggered by mechanical stimuli. Am. J. Physiol. 1997; 272: C1058-C1066.
Jiang Q. et al. Cystic fibrosis transmembrane conductance regulator-associated ATP release is controlled by a chloride sensor. J. Cell Biol. 1998; 143: 645-65.7
Scopus (87)
PubMed
Cotrina M.L. et al. Connexins regulate calcium signaling by controlling ATP release.
Proc. Natl. Acad. Sci. U. S. A. 1998; 95: 15735-15740.
Scopus (597)
PubMed
Trexler E.B. et al. Voltage gating and permeation in a gap junction hemichannel. Proc. Natl. Acad. Sci. U. S. A. 1996; 93: 5836-5841.
Scopus (244)
PubMed
Li H. et al. Properties and regulation of gap junctional hemichannels in the plasma membranes of cultured cells. J. Cell Biol. 1996; 134: 1019-1030.
Scopus (267)
PubMed
Cotrina M.L. et al. ATP-mediated glia signaling. J. Neurosci. 2000; 20: 2835-2844.
PubMed
Kandler K. Katz L.C. Neuronal coupling and uncoupling in the developing nervous system. Curr. Opin. Neurobiol. 1995; 5: 98-105.
Scopus (149)
PubMed
Mirsky R. Jessen K.R. The neurobiology of Schwann cells. Brain Pathol. 1999; 9: 293-311.
PubMed
Fitzgerald M. Spontaneous and evoked activity of fetal primary afferents in vivo. Nature. 1987; 326: 603-605.
Scopus (96)
PubMed
Wang D.J., Multiple signal transduction pathways lead to extracellular ATP-stimulated mitogenesis in mammalian cells: I. Involvement of protein kinase C-dependent and independent pathways. J. Cell Physiol. 1991; 146: 473-482.
Scopus (22)
PubMed
Neary J.T. et al. Mitogenic signaling by ATP/P2Y purinergic receptors in astrocytes: involvement of a calcium-independent protein kinase C, extracellular signal-regulated protein kinase pathway distinct from the phosphatidylinositol-specific phospholipase C/calcium pathway.
J. Neurosci. 1999; 19: 4211-4220.
PubMed
Gallo V. et al. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block.
J. Neurosci. 1996; 16: 2659-2670.
PubMed
Barres B.A., Raff M.C. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature. 1993; 361: 258-260.
Scopus (395)
PubMed
Grafe P. et al. Confocal calcium imaging reveals an ionotropic P2 nucleotide receptor in the paranodal membrane of rat Schwann cells.
J. Physiol. 1999; 515: 377-383.
Scopus (41)
PubMed
Mayer C. et al. Differences in the sensitivity to purinergic stimulation of myelinating and non-myelinating Schwann cells in peripheral human and rat nerve. Glia. 1998; 23: 374-382.
Scopus (36)
PubMed
Green A.C. et al. ATP acting on P2Y receptors triggers calcium mobilization in Schwann cells at the neuroelectrocyte junction in skate. Neuroscience. 1997; 80: 635-651.
Scopus (21)
PubMed
Fulton B.P. Gap junctions in the developing nervous system. Perspect. Dev. Neurobiol. 1995; 2: 327-334.
PubMed
Rathbone M.P. et al. The trophic effects of purines and purinergic signaling in pathologic reactions of astrocytes. Alzheimer Dis. Assoc. Disord. 1998; 12: S36-S45.
Scopus (39)
PubMed
Illes P. et al. Molecular mechanisms of microglial activation. B. Voltage- and purinoceptor- operated channels in microglia. Neurochem. Int. 1996; 29: 13-24.
Scopus (71)
PubMed
Stella N. et al. Interleukin-1 enhances the ATP-evoked release of arachidonic acid from mouse astrocytes. J. Neurosci. 1997; 17: 2939-2946.
PubMed
Franke H. et al. P2 receptor-mediated proliferative effects on astrocytes in vivo. Glia. 1999; 28: 190-200.
Scopus (99)
PubMed
Hindley S. et al. Stimulation of reactive astrogliosis in vivo by extracellular adenosine diphosphate or an adenosine A2 receptor agonist. J. Neurosci. Res. 1994; 38: 399- 406.
Scopus (81)
PubMed
Grafstein B. et al. Meningeal cells can communicate with astrocytes by calcium signaling.
Ann. Neurol. 2000; 47: 18-25
Scopus (35)
PubMed
Wieraszko A. et al. Stimulation-dependent release of adenosine triphosphate from hippocampal slices. Brain Res. 1989; 485: 244-250
Scopus (191)
PubMed
Gilmore S.A. Sims T.J. Glial cells and regeneration in the peripheral nervous system. in: Kettenmann H. , Ransom B.R. (Eds.) Neuroglia. Oxford University Press, ; 1995
Antioxidant reference
Jones, S. and Janardhanan, K.K. Antioxidant and antitumor activity of Ganoderma Lucidum (Curt. :Fr.) P. Karst.-Reishi (Aphyllophoromycetidea) from South India, Int. J. Med. Mushrooms, 2, 195-200, 2000.
Chinese University of Hong Kong, Ganodermic acids A, B, C, and D, lucidenic acid B, and ganodermanontriol are all powerful antioxidants.
The anti-androgen effect of ganoderol B isolated from the fruit body of Ganoderma lucidum. Liu J., Shimizu K., Konishi F. KumamotoS., Konda R., Bioorg Med Chem. 2007; 15 (14):4966-72.
Alzheimer reference
Aurifulria polyricha in processed and unprocessed from exhibited inhibitory activity against APP- cleaving enzyme (BACE1), which is responsible for releasing toxic β-amyloid peptide in the brain. Bennett L. Sheean P., Zabaras D., Head R., Int J Med Mushrooms. 2013; 15(3): 233-49.
Antagonizing beta-amyloid peptide neurotoxicity of the antiaging fungus Ganoderma lucidum. Lai CS., Yu Ms., Yuen WH., So KF., Zee SY., Chang RC. Brain Res. 2008;1190:215-24.
FNL Scientists and Collaborators Solve 3-D Structure of Key Protein in Alzheimer’s Disease | FNLCR Staging Researchers at the Frederick National Lab (FNL) have collaborated in solving the three-dimensional structure of a key protein in Alzheimer’s disease, providing new insight into the basic mechanisms that give rise to the devastating illness.